染色体微阵列分析技术在不明原因神经发育障碍性疾病患儿中的应用
2021-06-04闫冬梅王艳娟赵亚丽杨舒婷王志伟王雷雷
邵 晨,闫冬梅,王艳娟,赵亚丽,杨舒婷,王志伟,刘 双,仝 娇,王雷雷
神经发育障碍性疾病(neurodevelopmental disabilities,NDDs)通常是指由于异常的神经发育基础而导致的精神、心理、行为发育障碍,包括发育落后/智力障碍(developmental delay/intellectual disability,DD/ID)、学习障碍、自闭症症候群(autism spectrum disorders,ASD)等,影响超过15%的儿童[1]。DD/ID是指发育成熟以前(18岁以前)出现的认知和适应行为障碍,是儿科门诊较为常见的疾病之一,国外报道DD/ID的患病率为1%~3%[2],ASD是一组严重影响儿童健康、具有显著临床和病因异质性的常见的NDDs。
NDDs发病机制复杂,与环境、遗传等因素关系密切[3],其中遗传因素是主要因素之一。以ASD为例,一半以上的致病危险因素可归因于遗传学的变异;DD/ID的患儿中,有一半以上可以找到遗传学病因[4]。遗传性因素包括染色体数目和结构异常、单基因病、线粒体病、多基因和/或表观遗传异常等。据统计,染色体数目和结构异常占整个遗传因素的25%~30%[5]。拷贝数变异(copy number variations,CNVs)是由基因组发生重排而导致的,一般指长度为1 kb至数Mb的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。CNVs是导致DD/ID和ASD发生的重要病因,可以引起基因组与分子表型的异质性,导致疾病的发生。
近年来,随着基因组学技术的快速发展,NDDs遗传分子机制及诊断方法的研究取得了很大的进展[6]。染色体微阵列芯片分析(chromosomal microarray analysis,CMA)又称染色体基因芯片分析技术,能够检测出>100 kb的CNVs[7],在西方国家DD/ID、ASD等疾病的临床诊断中得到广泛应用[8-9]。相比于传统的染色体核型分析,CMA能够显著提高具有临床意义的CNVs的检出率,已成为DD/ID、ASD及多发畸形等疾病的一线实验诊断方法[10-11]。文献[12-13]报道显示,CMA在“原发性”智力障碍或者无法解释的发育迟缓、自闭症和先天性结构畸形病例中体现出了重大应用价值。但CMA技术应用于我国NDDs儿童诊断的临床实践起步较晚,目前我国CMA在NDDs儿童的临床分子诊断中的检测性能等信息不够完善,因此本研究对100例NDDs患儿进行CMA检测,针对检测结果进行分析,该研究将有助于扩充临床数据,为CMA在儿科疾病诊断中的广泛应用和遗传咨询提供参考。
1 资料与方法
1.1 一般资料 收集2016年3月至2018年8月于连云港市妇幼保健院就诊的NDDs患儿100例,其中男72例,女28例,年龄40 d至11岁,平均年龄3.31岁;ASD患儿55例,DD/ID患儿45例。所有患儿的父母均进行CMA检测前的遗传咨询并签署知情同意书,本研究经连云港市妇幼保健院医学伦理委员会审核批准。
1.2 纳入标准 (1)年龄0~18岁;(2)ID:依据韦氏智力测试(WISC-R及WPPST)IQ<70;DD:婴幼儿采用Gesell发育量表评测,DQ<75者诊断发育迟缓;ASD患儿为通过美国精神障碍诊断与统计手册(第5版)[14];(3)临床表现及实验室检查无代谢性病、单基因遗传变性病证据;(4)获得病人父母或其他监护人的知情同意,愿意参加本研究。
1.3 排除标准 (1)诊断不明确,或伴有其他神经系统器质性疾病;(2)细胞遗传学分析已明确病因的患儿,如21-三体、18-三体、13-三体综合征等;(3)临床资料不齐全和/或已诊断病因的患儿病例;(4)患有神经系统疾病的患儿,如脑瘫、癫痫、精神分裂症、Rett综合征等精神神经疾病及聋哑等。
1.4 研究方法
1.4.1 CMA检测 样本采集:利用EDTA抗凝采血管采集本研究中NDDs患儿外周血2 mL。采用提取试剂盒(QIAamp@DNA Blood Mini Kit)提取全基因组DNA,并于-20 ℃环境中储存备用。染色体微阵列分析检测将5 μL全基因组DNA用NspI酶随机消化为短片段,消化后产物末端补齐,连接上共同引物。将上述处理后样本DNA扩增为150~2 000 bp的片段,将扩增产物通过磁珠法纯化。将纯化产物通过片段化酶片段化为25~125 bp的片段,连接上生物素标记。将标记后产物与杂交液混合均匀并变性后加载于美国Affymetrix公司的CytoScan750K芯片(包括550 000个非多态性CNV探针和200 000个SNP探针),置于杂交仪中(Genechip hybridization oven 645)50 ℃,60 r/min,杂交16~18 h后,置于洗涤工作站(GeneChip@FluidiesStation450Dx v.2)洗涤并染色。将洗涤后芯片置于扫描仪(GeneChip@Scanner 3000Dx v.2withAutoLoader)中扫描,获取数据并加载于配套ChASv3.0软件后利用相关的生物信息学方法分析检测结果。
1.4.2 CMA数据分析及结果判读 选择芯片识别的≥200 kb的微缺失和≥500 kb的微重复且可信度≥90%的片段利用各种生物信息学数据库进行分析。相关数据库主要包括人类孟德尔遗传数据库(Online Mendelian Inheritance In Man,OMIM,https://www.omim.org/)、国际公共良性CNVs数据库(Database of Genomic Variants,DGVs,http://dgv.tcag.ca/dgv/app/home)、人类基因组变异和表型数据库(Database of genomic variation and Phenotype in Humans using Ensembl Resources,DECIPHER,http://decipher.Sanger.Ac.uk/),人类遗传学细胞遗传学微阵列委员会在线数据库(International Standard Cytogenomic Array Consortium,ISCA,http://www.iscaconsortium.org/)、美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI,https://www.ncbi.nlm.nih.gov/ pubmed/)等。根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)对CNVs解释的标准和指南[10],按照临床意义将所有检测到的非多态性CNVs分成以下5类:(1)致病性CNVs;(2)可能为致病性CNVs;(3)临床意义未知CNVs;(4)可能良性CNVs;(5)良性CNVs。
2 结果
2.1 CMA检测出的CNVs 利用750K芯片对100例DD/ID及ASD患儿进行了检测,结果显示22例(22.00%,22/100)患儿检测到染色体拷贝数变异,携带病理性及可能致病CNVs患儿14例(14/100,14.00%),共涉及17个致病性及可能致病性CNVs,包括14个微缺失(1q21.1q21.2、6p22.3、7q11.23、7q11.23、7q31.1、8p23.3p23.1、9q34.3、10q26.13q26.3、15q11.2q13.2、15q11.2q13.1、15q11.2q13.1、Xp22.32p22.31、Xp22.33p11.23、Xq21.1q28);3个微重复(2q36.3q37.3、9q34.12q34.3、8q24.23q24.3)(见表1),检出率达14.00%。携带临床意义未知CNVs的患儿有8例(8/100,8.00%)。78例患儿染色体未见明显异常,所有样本的检测成功率为100%。

表1 CMA检测结果[n;百分率(%)]
对45例DD/ID患儿进行CMA检测发现12例病理性或可能致病性CNVs,检出率为26.67%,其中男3例,女9例。12例DD/ID患儿中检测出的14个病理性及可能致病性CNVs,其中12个染色体微缺失,2个染色体微重复(见表2)。同时在对55例ASD患儿CMA检测中发现2例病理性及可能致病性CNVs,检出率为3.64%,其中男1例,女1例。在2例ASD患儿中检测出的3个病理性CNVs包含2个染色体微缺失,1个微重复(见表3);病理性及可能致病性CNVs中发现与神经发育相关的OMIM基因有MCPH1、CLN8、TRAPPC9、GPAA1、PUF60等。

表2 DD/ID病人中CMA检测结果为病理性及可能致病CNVs

表3 ASD病人中CMA检测结果为病理性及可能致病CNVs
对8例临床意义未知病例进行CMA检测发现10个临床意义未知CNVs,包括4个微缺失(3p22.2、5q33.3、7q34、22q13.2),6个微重复(2q21.3q22.1、16q24.3、19q13.42q13.43、Xq11.2q12、Xq26.2、Yq11.223q11.23)(见表4)。在这些患儿中发现与神经发育相关的OMIM基因,分别是BRAF、ANKRD11,FANCA、AMER1、GPC3。

表4 临床意义未知CNVs
2.2 CNVs染色体分布 检测出2例ASD患儿的病理性CNVs,分别分布在8号、X染色体上,其中1例患儿在8号染色体上同时发现缺失片段及重复片段。12例DD/ID患儿检测出的病理性及可能致病性CNVs分布在1号、2号、6号、7号、9号、10号、15号、X号染色体上。其中3例患儿病理性及可能致病性CNVs分布在15号染色体上,3例患儿病理性及可能致病性CNVs分布在7号染色体上。2例患儿病理性及可能致病性CNVs分布在9号染色体上,2例患儿病理性及可能致病性CNVs分布在X染色体上。1例患儿X染色体上发现2个缺失片段。在8例患儿检测出的临床意义未知的CNVs分布在2号、3号、5号、7号、16号、19号、22号、X、Y染色体上。其中1例患儿分别在3号、5号、7号染色体上发现缺失片段。2例患儿临床意义未知CNVs分布在X染色体(见图1)。

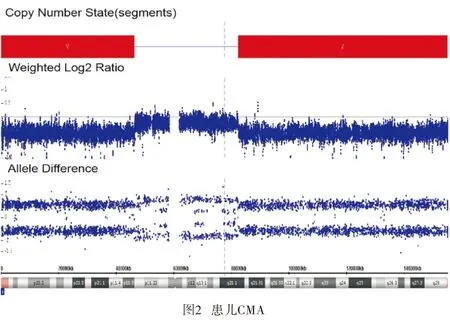
1例诊断为DD的患儿,CMA技术检测的致病性CNVs分析图见图2,该片段有两处缺失,分别是:arr[hg19]Xp22.33p11.23(168,551 -46,489,686)x1,大小为46.3 Mb,包含OMIM基因数156个;arr[hg19]Xq21.1q28 (82,441,085-155,233,098)×1,大小为72.79Mb,包含OMIM基因数304个,患儿为女孩,7个月大,表现发育迟缓。
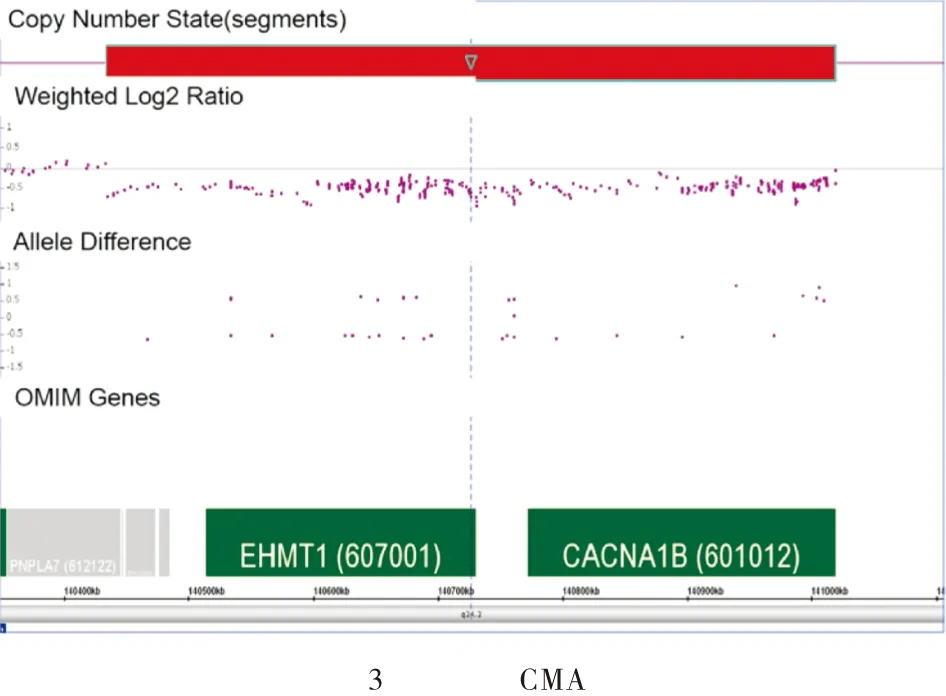
1例诊断为DD的9q34.3微缺失综合征患儿,CMA技术检测的致病性CNVs分析图见图3,该片段有一处缺失:arr[hg19] 9q34.3(140,434,785-141,018,648)x1,大小为0.58 Mb,包含OMIM基因数6个。患儿为女孩,14个月大,表现发育迟缓。
3 讨论
随着二胎政策的放开,高龄产妇的增多,孕期暴露风险因素的可变性,越来越多的家庭迎来了第二个宝宝的同时NDDs儿童的数量也在逐年增长,NDDs的发病率因疾病类型不同差异较大,但整体受累人群比例较高。以ASD为例,全球人口发病率约为0.6%[15],因我国人口众多,NDDs对我国儿童的健康带来巨大的威胁,且至今无有效的治疗方法,不仅给家庭带来经济负担和心理负担,也给社会带来沉重负担,同时严重影响人口素质。CNVs是导致DD/ID及ASD等NDDs发病的遗传学因素之一,而目前西方发达国家对CNVs的研究主要采用CMA技术,该技术已成为DD/ID、多发畸形及ASD等疾病的一线诊断实验[10],但CMA技术在我国起步较晚,本研究旨在探讨CMA技术在发掘NDDs遗传学因素中的应用,同时为本地区建立一套完整的基因诊断路径,为以后儿科遗传疾病的遗传分析提供指导。
染色体异常和基因组相关的微缺失微重复是NDDs患儿重要的遗传学病因,常染色体显性遗传占智力障碍或全面发育迟缓比例为13%~39%[2],新生突变是导致重度智力障碍或全面发育迟缓的重要病因[16]。常染色体隐性遗传占智力障碍或全面发育迟缓比例为10%~20%[17]。本研究显示,CMA对ASD患儿的分子诊断率为3.64%,CMA对DD/ID患儿的分子诊断率为26.67%,诊断水平高于有研究[18]已报道的CMA对DD/ID分子诊断率14.7%;低于CMA对ASD分子诊断率12%,高于刘欣等[3]已报道的CMA对DD/ID分子诊断率26.3%。分析原因主要为两个方面,一是纳入标准不同,刘欣等的纳入标准为诊断为ASD、DD/ID、学习障碍、注意力缺失/多动症(attention deficit hyperactivity disorder,ADHD)的患儿及部分伴有面容异常或多发畸形的患儿,纳入病种比本研究多样。二是地域之间的差异。该结果对本地区的NDDs患儿临床分子诊断,干预方面具有指导意义。
本研究在101例NDDs病人中检出的18个病理性及可能致病性CNVs中包括3个已知的NDDs致病性热点区域,即1q21.1q21.2、15q11.2q13.1、7q11.23,与国外已报道的情况[18]类似,说明这些已鉴定的热点变异同样在我国儿童NDDs的发生中发挥重要作用。对检测结果为临床意义未知CNVs的8个病例中,4例患儿发现了与已报道的神经发育相关的基因变异,分别是:BRAF、ANKRD11、FANCA、AMER1、GPC3。WU等[19]报道Noonan综合征是一种常染色体显性遗传病,主要临床表现为先天性心脏病、身材矮小、特殊面容、发育迟缓、凝血功能障碍、骨骼畸形等,BRAF基因新生突变导致Noonan综合征的发生。SACHAROW等[20]报道ANKRD11 基因突变所致KBG 综合征,以广泛发育迟缓(语言发育迟缓尤为突出)、颅面异常、上颌中切牙过大和骨骼异常为特征的罕见常染色体显性遗传性疾病。值得注意的是,人种间基因组的差异可能导致同一致病性位点在不同人群中的发生频率出现明显差异,比如Phelan-McDermid综合征相关位点22q13微缺失在中国ID患儿中的发生明显高于西方(1.7%和0.24%)[21]。这一现象表明建立我国患儿NDDs致病性CNVs谱系数据库对有效开展相关领域研究的重要性和必要性。
我们对DD/ID病人检出的14个致病性CNVs分析发现与XU等[18]已报道的一些经典的综合征位点如1q21.1q21.2、15q11.2q13.1、7q11.23相吻合。另外,我们还发现1例X染色体异常的发育迟缓患儿;人类全基因组有25 000个功能基因(目前仍在增加中),其中X染色体上有829个,仅占人类基因组基因的3.3%,然而10%~15%的精神发育迟滞与X染色体基因/基因组变异有关[22],所以本例中的发育迟缓患儿可能与其染色体异常密切相关。另外我们还发现1例9q34.3微缺失综合征,研究[23]表明9q34.3微缺失综合征主要临床表型包括智力低下、发育迟缓、语言发育障碍、肌张力低下、特殊面容(如眼距增宽、鼻孔前倾、浓眉),部分病人可能出现新生儿喂养困难、食管反流、癫痫、心脏畸形及肥胖表型[23]。本研究中的9q34.3微缺失综合征患儿有相似症状。另外9号染色体上不同区段的重复临床表型也会不同,9p22.1-p22.3区域重复与特殊面容有关,9p21.2-21.3区域重复与语言发育落后有关,9p22.3-24区域的重复与智力低下有关[24-25]。
ASD病因复杂,近年来研究[26-27]发现,CNVs在ASD病因中的构成比为10%~20%。静进等[28]研究发现ASD病人染色体异常包括2q24缺失、3p14缺失和重复、1p36缺失、3q27~3q28缺失、7p21缺失等,涉及的易感基因包括MET、RELN、FOXP2、NRP2、PTEN、TsC1/Tsc2等。另外有多项研究发现EN2、RELN、NRP2、OXTR、GluR6和FOXP2基因的突变或多态性可能与中国人的ASD有关[29]。该研究中对ASD患儿检测出2例染色体异常包含3个CNVs分别是:8p23.3p23.1缺失、8q24.23q24.3重复、Xp22.32p22.31缺失。其中1例患儿在8号染色体上同时发现缺失片段及重复片段。与神经发育相关的OMIM基因有:MCPH1、CLN8、TRAPPC9、GPAA1、PUF60、NLGN4。DUERINCKX等[30]的研究表明常染色体隐性智力缺陷(Autosomal recessive intellectual disability,ARID)是智力缺陷的非常不同的亚组,而TRAPPC9和MCPH1的每个突变都会导致一种ARID。LOW等[31]研究发现12例病人中观察到的临床和分子数据表明PUF60的变异导致以身材矮小为特征的综合征,发育迟缓、面部畸形等。还有学者[32]报道了5个新发的PUF60变异与偶发的ID患儿常见先天性异常相关的。基于以上研究,我们认为TRAPPC9、MCPH1和PUF60可能是导致ASD发病的原因,关于他们的致病机制需要进一步研究。
