固相萃取-同位素稀释-UPLC-MS/MS法检测茶叶中百草枯和敌草快残留量
2021-05-25陈健豪
陈健豪
(福建省产品质量检验研究院,福建 福州 350002)
百草枯(1,1'-二甲基-4,4'-联吡啶阳离子盐)和敌草快(1,1'-亚乙基-2,2'-联吡啶二溴盐)均为快速灭生性除草剂,具有触杀作用和一定内吸作用,能迅速被植物绿色组织吸收,使其枯死。百草枯与敌草快在土壤中迅速与土壤结合而钝化,在农业上广泛用于茶园、果园中的杂草去除。但是,百草枯对人毒性极大,且无特效解毒药,口服中毒死亡率极高[1],已被20多个国家禁止或者严格限制使用。中国自2014年7月1日起撤销了百草枯水剂登记和生产许可、停止生产,但保留母药生产企业水剂出口境外登记、允许专供出口生产,2016年7月1日起停止水剂在国内销售和使用[2]。
我国GB 2763—2019《食品安全国家标准 食品中农药最大残留限量》[3]规定茶叶中百草枯的残留限量为0.2 mg/kg,但对茶叶中敌草快的限量未做规定,而实际上在茶叶种植管理中百草枯和敌草快这2种除草剂都不允许使用。按国标GB 2763—2019规定,百草枯检测方法为SN/T 0293—2014[4],敌草快检测方法为GB/T 5009.221—2008[5]及SN/T 0293—2014。SN/T 0293 —2014标准方法适用于蔬菜、水果等机体,由于茶叶基质非常复杂,在茶叶检测中发现该方法存在检测灵敏度极低、回收率差等缺点。GB/T 5009.221—2008标准在检测敌草快时采用气相色谱法,由于敌草快为强极性不挥发的物质,因此,需要复杂的衍生和萃取步骤,检测效率极低。目前,对百草枯和敌草快的检测方法主要采用液相色谱-串联质谱联用法[6~11],但现有方法在检测茶叶时往往都存在前处理方法复杂、不适用、离子抑制强烈、回收率差和定性、定量不准确等问题,无法达到快速、灵敏检测的目的。因此,本方法在已有文献方法的基础上,对茶叶的前处理方法、分离检测条件及检测影响因素等进行了详细的研究。
1 试验部分
1.1 试验仪器
超高效液相色谱仪Waters Acquity UPLC系统配Quattro Premier XE串联四极杆质谱仪(美国沃特世公司);20管固相萃取装置(美国安捷伦公司);冷冻高速离心机(美国贝克曼公司,Avanti J-E);超声清洗机(鼎泰(湖北)生化科技设备制造有限公司,DTC-27J); Milli-Q超纯水纯化系统(美国Millipore公司,Advantage);分析天平(赛多利斯北京有限公司,BSA224S);涡旋混合器(德国IKA公司,MS 3 basic)。
1.2 试剂和材料
水为超纯水;甲醇(色谱纯,山东禹王);乙腈(色谱纯,山东禹王);甲酸(GR,德国默克);盐酸(AR)、乙酸铵(GR)均为国药集团化学试剂有限公司生产;标准品:二氯百草枯标准品(99.9%,美国sigma公司);敌草快二溴盐标准品(99.0%,德国Dr.Ehrenstorfer公司);百草枯-D6二碘化物(99.8%,上海甄准生物科技有限公司);敌草快-D4同位素内标(93.7%,德国Dr.Ehrenstorfer公司);固相萃取柱:PCX 60 mg/3 mL(博纳艾杰尔科技有限公司),使用前分别用2 mL甲醇和2 mL 0.5%甲酸溶液活化平衡。
5 mol/L 乙酸铵:称取38.5 g乙酸铵,加水溶解稀释到100 mL。
0.5 mol/L 盐酸:移取20.0 mL盐酸,加水稀释到500 mL。
0.5 mol/L 盐酸∶甲醇=2∶8提取液:取200 mL 0.5 mol/L盐酸加800 mL甲醇,混匀。
50% 乙腈(含2%甲酸):250 mL乙腈加250 mL水,再加10 mL甲酸,混匀。
淋洗液:取1.0 mL浓度为5 mol/L乙酸铵溶液,加入4.0 mL水、4.0 mL甲醇和1.0 mL甲酸,混匀。
洗脱液:取4.0 mL浓度为5 mol/L乙酸铵溶液,加入1.0 mL水、4.0 mL甲醇和2.0 mL甲酸,混匀。
标准溶液和内标溶液配制:分别准确称取适量百草枯和敌草快标准品,用0.1 mol/L盐酸∶乙腈=1∶1溶液溶解配成百草枯和敌草快浓度约为500 μg/mL的标准储备溶液,再取适量储备液用0.1 mol/L盐酸∶乙腈=1∶1溶液稀释成约1 μg/mL的标准中间液,置于4 ℃冰箱中保存。
分别称取适量的百草枯-D6和敌草快-D4标准品,用0.1 mol/L盐酸∶乙腈=1∶1溶液溶解配制成约500 μg/mL的内标标准储备溶液,再取适量内标储备液用0.1 mol/L盐酸∶乙腈=1∶1溶液稀释成约10 μ g/mL的内标标准中间液,置于4 ℃冰箱中保存;移取适量百草枯和敌草快标准中间溶液,准确加入20 μL同位素内标中间溶液,用洗脱液稀释定容至2.0 mL,混匀,配成百草枯和敌草快浓度约为5~250 ng/mL,内标浓度均为100 ng/mL的标准系列,将标准溶液用移液枪吸入塑料内衬管中,置于2 mL样品瓶中进行UPLC-MS/MS分析。
1.3 样品处理
称取5 g(精确到1 mg)粉碎茶叶样品于50 ml具塞塑料离心管中,加入50 μL内标中间液,再加入25.0 mL 0.5 mol/L盐酸∶甲醇=2∶8提取液,涡旋振荡混匀,超声波振荡提取30 min,10000 r/min 离心3 min。用移液枪移取5.0 mL上清液加入活化后的PCX 60 mg/3 mL固相萃取柱中,流速控制在约1 mL/min,待样液流尽后,用2 mL50%乙腈(含2%甲酸)、2 mL甲醇和1.0 mL淋洗液依次淋洗,真空抽干,再用1.0 mL洗脱液洗脱于10 mL塑料离心管中,涡旋混匀,移取300 μL洗脱液于塑料内衬管中,置于2 mL样品瓶中进行UPLC-MS/MS分析。
1.4 UPLC-MS/MS 分析条件:
1.4.1 色谱分析条件

表1 特征离子及其他质谱参数
色谱柱:资生堂,Capcell PAK,ST,5 μm,150 mm×2.0 mm;柱温:35℃;进样量:10 μL;流速:0.40 mL/ min;
流动相及梯度:A为20 mmol/L乙酸铵(含0.4%甲酸),B为乙腈,0—1.0 min,30%—50% A;1.0—5.0 min,50% A;5.0—6.5 min,90% A;6.5—8.5 min,30% A。
1.4.2 质谱分析条件
电离源:电喷雾正离子模式;毛细管电压:4.00 kV;源温度:120 ℃;脱溶剂气温度:400 ℃;脱溶剂气流量:700 L/h;碰撞气:氩气,0.40 mL/min;电子倍增管电压650 V;检测方式:多反应监测[multi reaction monitoring(MRM)]模式;特征离子及其他质谱参数见表1。
2 结果与讨论
2.1 质谱条件优化
分别取浓度约为1 μg/mL的百草枯和敌草快标准溶液及内标溶液,由注射泵注入质谱仪扫描进行母离子和子离子的扫描,扫描结果发现在ESI(+)模式下,百草枯同时存在响应值相近m/z为186和185的母离子,敌草快母离子m/z为183,百草枯-D6 母离子m/z为192,敌草快-D4母离子m/z为186,通过优化锥孔电压等参数得到扫描图(图1)。
对母离子进行子离子扫描,由于百草枯m/z为186和185的母离子都只有一个丰度较大的子离子,其余子离子的响应丰度值相比都要小20倍以上,因此,百草枯检测选择186>171离子对为定量离子,选择185>170离子对为定性离子,通过优化碰撞能量等参数得到百草枯和敌草快及内标的子离子扫描图(图2),最终确定百草枯和敌草快的定量和定性离子对及内标离子对相应的质谱参数如“1.4.2”。

图1 百草枯和敌草快及内标的一级质谱分析图
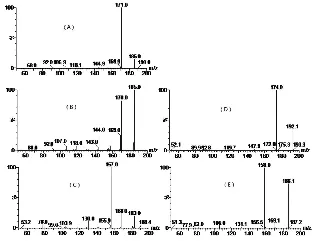
图2 百草枯和敌草快及内标的二级质谱分析图
2.2 色谱条件优化
本方法采用了超高效液相色谱仪,该仪器具有较小体积,且在低流速条件下能够对梯度变化有很快的响应。
由于百草枯和敌草快都是强极性的物质,在C18色谱柱上基本没有保留,因此,采用适合极性物质分离的亲水性HILIC色谱柱,实验对比了Waters公司的1.7 μm,100 mm×2.1 mm HILIC色谱柱和资生堂公司的Capcell PAK,ST,5 μm,150 mm×2.0 mm色谱柱;同时,比较了0.1%甲酸/乙腈体系、0.5%甲酸/乙腈体系、10 mmol/L乙酸铵/乙腈体系和20 mmol/L乙酸铵/乙腈体系作为流动相的分离效果。实验结果发现,甲酸/乙腈体系下百草枯和敌草快色谱峰型差且灵敏度低,乙酸铵/乙腈体系能有效改善色谱峰型并提高灵敏度,提高乙酸铵浓度有利于改善峰型,增加响应强度,但浓度太高的乙酸铵缓冲盐容易造成离子源的快速污染,反而影响检测的灵敏度,因此,本实验采用20 mmol/L乙酸铵/乙腈体系做流动相。
针对流动相酸度的影响进行了实验,结果表明提高酸度能加快百草枯和敌草快的出峰,有效减小峰展宽,提高峰响应值,流动相中加入0.4%甲酸具有最好的分离效果和灵敏度。

图3 百草枯和敌草快标准品(A)和空白茶叶样品(B)总离子流图(TIC)和MRM谱图
在色谱柱选择上,虽然Waters公司的1.7 μm超高效色谱柱具有更好的峰型和灵敏度,但该柱难以将百草枯和敌草快分离,而百草枯的定性离子与敌草快存在干扰峰,必须将两物质分离达到基线分离,实验发现资生堂公司的Capcell PAK,ST色谱柱能将其有效分离,经优化后的色谱分离图见图3。
2.3 前处理条件优化
SN/T 0293 —2014标准所规定的检测方法适用范围中不包含茶叶,而实验过程中也发现无法正常检测。这是由于SN/T 0293—2014采用的固相萃取柱PWCX小柱为弱阳离子交换柱,对百草枯和敌草快的吸附不够强,很容易由于茶叶中的大量杂质竞争吸附而解吸,同时样品中大量的杂质也造成离子抑制强烈,因此该净化方法完全不适用于茶叶样品。而本研究发现PCX强阳离子交换柱对百草枯和敌草快有强烈的吸附,通用的PCX柱洗脱剂5%氨水/甲醇溶液无法将其洗脱,因此,可以将大量的样品杂质通过50%乙腈(含2%甲酸)和甲醇等各种淋洗剂去除掉,最后采用较高浓度的酸性乙酸铵/甲醇洗脱溶液1 ml即可有效洗脱百草枯和敌草快。
提取溶液优化。百草枯和敌草快均极易溶于水,但茶叶吸水后膨胀非常厉害,并且,高速离心后的上清液中仍然含有大量的胶质等悬浮物,在过柱净化时极易造成堵塞,严重影响净化效果和回收率。研究发现增加提取液中甲醇比例能够有效解决溶胀和堵塞问题,同时,在酸性条件下有利于百草枯和敌草快的电离及在PCX柱上的吸附,因此,实验最终采用0.5 N盐酸∶甲醇=2∶8作为样品提取溶液。此外,实验发现玻璃材质对百草枯和敌草快具有较强的吸附作用,低浓度时甚至被完全吸附,因此,样品提取、前处理及进样溶液均需采用塑料制备器具与容器。
2.4 线性范围和检测限
本方法采用内标法定量,标准系列只需用样品洗脱溶液配制即可。在5~250 ng/mL浓度范围内,以标准工作溶液浓度为横坐标(X),以百草枯和敌草快定量离子峰面积除以内标峰面积的值与内标浓度的乘积为纵坐标(Y),绘制标准工作曲线,在该浓度范围内百草枯和敌草快均具有良好的线性关系,由10 ng/mL样品加标溶液的峰高与噪音的信噪比计算得到茶叶基质中的方法检测限(3倍噪音)和定量限(10倍噪音)如表2。用标准工作曲线对试样中待测组分进行内标法定量,试样溶液的响应值应在标准工作曲线的线性范围内。

表2 百草枯和敌草快的线性方程与检测限
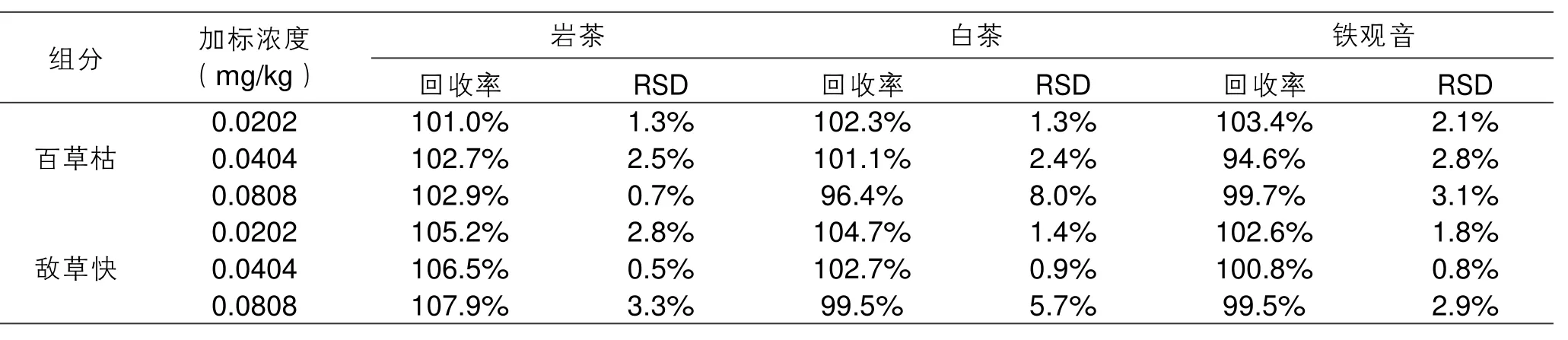
表3 不同茶叶中百草枯和敌草快的加标回收率和精密度(n=6)
2.5 回收率和精密度试验
取福建盛产的岩茶、白茶和铁观音3种空白茶叶样品进行3个水平的标准添加试验,每个添加量做6个平行试验,试验结果见表3。该方法对百草枯的回收率在94.6%~103.4%之间,精密度在0.7%~8.0%之间;对敌草快的回收率在99.5%~107.9%之间,精密度在0.5%~5.7%之间。可见该方法对百草枯和敌草快均具有良好的回收率和精密度,适合茶叶的检测。
3 实际样品检测
采集福建省内茶叶企业生产的岩茶、茉莉花茶、铁观音茶、红茶、白茶和绿茶等茶叶样品100个批次,采用本方法进行检测,检测结果敌草快均未检出,共有16个样品检出了百草枯,阳性样品检测结果见表4。
百草枯检出率为16%,其中最高检出含量为0.08 mg/kg,均在国标GB 2763—2019《食品安全国家标准 食品中农药最大残留限量》规定的限量0.2 mg/kg允许范围内,可见,在国家禁止百草枯农药使用以后,基本上已经杜绝了百草枯在茶叶种植中的使用,但由于百草枯与土壤结合牢固并会不断释放出微量的百草枯[12],因此,前期有使用过百草枯的茶园仍会在一段时间内残留少量的百草枯。

表4 福建省内茶企样品中百草枯阳性样品检测结果(n=3)
4 结论
本文建立了茶叶中百草枯和敌草快的提取、净化方法和同位素稀释-UPLC-MS/MS检测方法。茶叶样品经高有机相酸性提取液提取、离心后,使用PCX强阳离子交换小柱固相萃取净化,去除了茶叶中大量的干扰物质,降低了离子抑制的干扰,采用资生堂Capcell PAK,ST亲水色谱柱分离后UPLCMS/MS检测,内标法定量。该方法定性、定量准确,线性范围宽,灵敏度高,能很好地应用于茶叶中百草枯和敌草快的检测,填补了国标方法对茶叶检测的难题,为相关国家标准的修订提供了有力的技术支撑。
