儿童嘌呤核苷磷酸化酶缺乏症1 例报告并文献复习
2021-05-24钟志娟吉训琦陈玉雯冯小伟
钟志娟 吉训琦 陈玉雯 冯小伟
海南省人民医院海南医学院附属海南医院儿科(海南海口 571127)
嘌呤核苷磷酸化酶缺乏症(purine nucleoside phosphorylase def iciency,PNPD)是一种罕见的原发性免疫缺陷病,国外文献报道共78例,国内尚无相关报道。PNPD临床特征主要包括反复感染、自身免疫性疾病、神经功能异常等。本文报告海南省人民医院儿科收治的1 例PNPD 患儿的临床资料,并进行相关文献复习。
1 临床资料
患儿,男,1 岁9 月龄,因面色苍白2 月余,发热、咳嗽伴解浓茶样尿1 天于2019 年7 月入院。患儿入院前2 个月出现发热伴浓茶样尿、贫血,当地医院诊断“自身免疫性溶血性贫血”,予输注丙种球蛋白 1 g/(kg·d)×2天、甲基泼尼松龙 10 mg/(kg·d)×3天,并予输注洗涤红细胞纠正贫血、碱化血液等治疗,症状缓解,出院后口服甲泼尼龙8 mg/d。入院前1天患儿出现发热、咳嗽,解浓茶样尿。患儿入院前1年有7次呼吸道感染史。患儿为G2P2,足月顺产,出生体质量 3.1 kg,无缺氧窒息史。6个月抬头,8个月翻身。在儿童医院诊断“运动发育迟缓”,定期康复训练,现可扶站,不能独立行走。父母非近亲结婚,父母及5 岁姐姐身体健康。入院体格检查:脉搏 114 次/min,呼吸 30次/min,身高79 cm(<P3),体质量9.5 kg(P3~P15),口唇苍白;双肺呼吸音粗,心、腹无异常;四肢肌力及肌张力无异常。实验室检查:血常规白细胞4.65×109/L,淋巴细胞绝对值0.91×109/L,红细胞2.84×1012/L,血红蛋白88 g/L,血小板461×109/L,网织红细胞11.35 %;尿酸85 μmol/L;Coombs 试验间接(+),直接(++++);酸溶血试验阴性;抗核双链单链 组合、ENA多肽及抗中性粒细胞胞浆抗体均阴性;铁蛋白734.00 ng/mL,血清铁75.40 μmol/L,铁饱和度99.00 %,总铁结合力76.00 μmol/L;HIV抗体阴性;IgA 0.8 g/L,IgG 10.62 g/L,IgM 6.29 g/L,补体 C3 1.04 g/L,补体C4 0.26 g/L。骨髓常规示骨髓增生明显活跃,粒系占53.5%,红系增高占39.0%,巨核细胞、血小板不少。诊断为“自身免疫性溶血,支气管炎”,予甲基泼尼松龙及头孢美唑治疗好转后出院,继续口服甲泼尼龙 8 mg/d。
患儿出院后10天,再次因发热、咳嗽伴浓茶样尿2 天入院。查血常规白细胞1.40×109/L,中性粒细胞绝对值0.3×109/L,淋巴细胞绝对值0.6×109/L,红细胞3.32×1012/L,血红蛋白109 g/L,血小板24×109/L;尿酸29 μmol/L;血小板特异性和组织相关性(HLA)抗体阴性;总T细胞(CD3+)55.2%,T辅助/诱导细胞(CD4+)4.3%,T抑制/毒性细胞(CD8+)13.5%,总B 细胞10.8%,NK 细胞42.6%,CD 4+/ CD8+T 0.32。胸部CT示支气管肺炎,胸腺发育不良。
因患儿诊断不明,不能除外遗传性免疫缺陷病或血液病,经医学伦理审核以及患儿父母知情同意行基因检测。抽取患儿及其姐姐、父母各2 mL 外周血 (EDTA 抗凝),送武汉康圣达医学检验所行遗传性血液病和免疫缺陷病基因外显子DNA 检测;并结合患儿临床特征、突变位点正常人频率、突变类型等,采用Sanger测序方法验证患儿及其他家庭成员相关的基因突变位点。结果发现患儿PNP基因存在纯合错义变异c.722T>C:p.I241T;患儿姐姐、父母均为杂合携带者(图1)。查询国际上千人数据库证实为新发现的变异,使用SIFT、PolyPhen2、LRT、Mutation Taster软件对变异的致病性进行评估,该变异为临床意义不明确的遗传学改变。患儿确诊为PNPD。
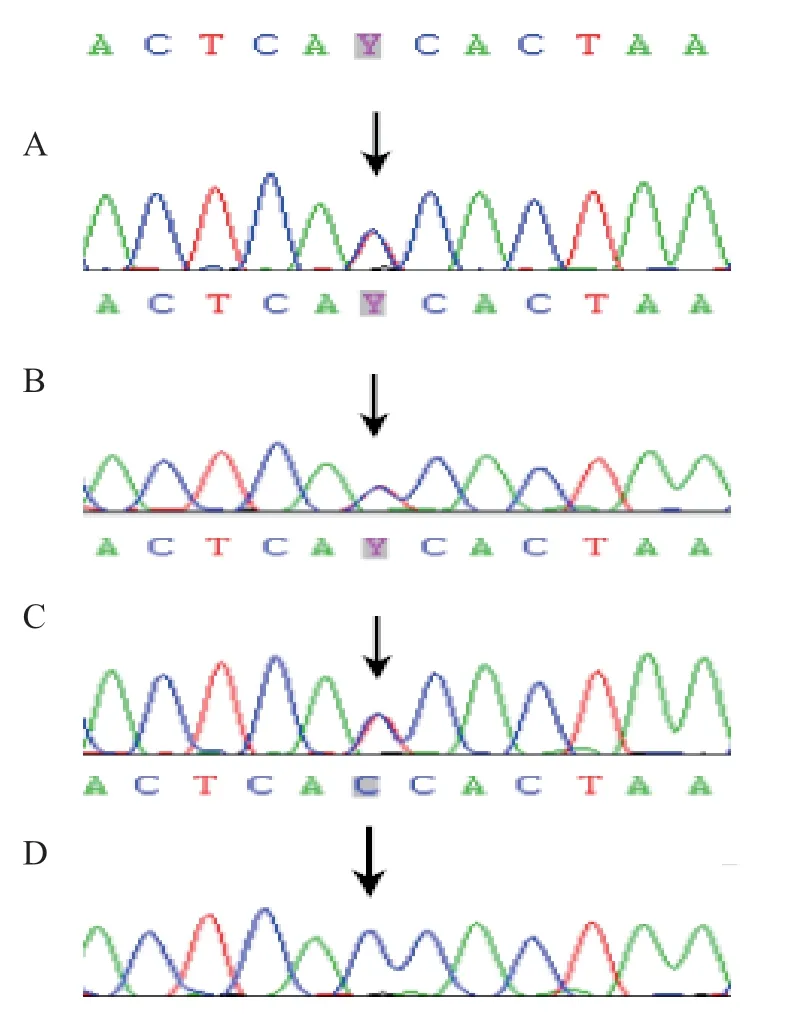
图1 患儿及家人PNP 基因 Sanger 测序图
患儿2岁3月龄时,因鼠伤寒沙门菌败血症、鼠伤寒沙门菌肠炎、肺曲霉菌病、肝脓肿等严重感染死于多脏器衰竭。
2 讨论
PNPD 由PNP基因变异导致嘌呤核苷磷酸化酶(purine nucleoside phosphorylase,PNP)缺乏所致,是常染色体隐性遗传性疾病。PNP基因位于染色体14 q 13.1 上,包含6 个外显子,编码由289 个氨基酸组成的PNP[1]。PNP 在人体大多数细胞中均有表达,但不同组织的PNP 水平不同,以淋巴组织表达最高。PNP 的功能是催化鸟苷转化为鸟嘌呤和催化次黄嘌呤,核苷转化为次黄嘌呤,最终生成尿酸。当PNP 缺乏时,血中鸟苷和次黄嘌呤增多,可导致淋巴细胞中脱氧三磷酸鸟苷(deoxyguanosine triphosphate,dGTP)含量升高,胸腺细胞中的dGTP也大量堆积,dGTP可抑制核糖核酸还原酶,该酶为DNA 合成的促进剂,故dGTP堆积抑制淋巴细胞合成DNA,使淋巴细胞增殖受限。由于B 淋巴细胞对PNP 缺乏有一定耐受能力,而T 淋巴细胞对PNP 缺乏敏感,故PNP 缺乏以细胞免疫缺陷为主,且Ts/Te 受PNP 缺乏的影响比TH/TI所受的更严重[2]。因此PNP缺乏者外周血中T细胞 数量显著减少,血液Ig 水平、B 细胞和浆细胞数可正常,血尿酸及PNP 酶水平低,影像学上可表现为胸腺发育不良。
复习文献,目前已报道的PNPD共78例[1,3-6],发病无性别差异,多发病于出生6~12 个月的婴儿,且随年龄增长,病情逐渐加重。在79例(含本例)PNPD患儿中54例表现为反复的病毒、细菌和真菌感染,其中10例有严重甚至致命的水痘病毒感染[1,4];52例有神经功能障碍,包括共济失调、不平衡、发育迟缓、高/低张力、痉挛性轻瘫、行为问题和智力低下;22例有自身免疫性疾病,最常见溶血性贫血有17例,另有自身免疫性血小板减少2例、系统性红斑狼疮2例、硬化性胆管炎3 例、干燥伴多发性硬化1 例和关节炎1 例。其中至少有7例在PNPD诊断前就诊断出自身免疫性疾病[4],6例继发肿瘤性疾病。57例患儿行血常规及淋巴细胞亚群分析,均有外周血淋巴细胞减少及T细胞减少,其中27例T细胞免疫低下同时B细胞免疫无异常,27例合并B细胞免疫缺陷;55例患儿行尿酸检测,45 例降低,10 例无异常(其中2 例采样前有明确输血史[1,4]);65例患儿行PNP活性测定中63例PNP低下,2例输血后查PNP酶1例正常、另1例接近正常[3-4]。51例患儿行基因分析,36例为纯合变异、15例为复合杂合变异;发现42 个独特的变异,35 个为错义变异,一些外显子跳跃、移码变异及小片段缺失亦有报道,反映了酶对双等位基因轻度变异的不耐受性。
PNPD 诊断主要依赖于临床特征、免疫学特点及基因测序分析。低尿酸水平是诊断的另一个辅助手段,PNP 酶活性或基因分析可作出明确诊断[3]。液相色谱-串联质谱法可用于新生儿及高危人群筛查[7]。本例患儿有运动发育迟缓、反复呼吸道感染、免疫性血细胞减少、淋巴细胞减少、T细胞免疫缺陷、低尿酸水平、胸腺发育不良及PNP基因的c.722T>C纯合变异,故PNPD诊断明确。
PNPD 治疗主要是积极预防和控制感染。合并低丙种球蛋白血症或疫苗抗原抗体应答障碍者可定期静脉输注丙种球蛋白(intravenous immunoglobulin,IVIG);合并自身免疫性疾病时,IVIG 冲击联合激素或其他免疫抑制剂有助于控制病情;严重溶血性贫血予输血支持治疗,血制品输注前需进行放射照射以减少移植物抗宿主病。造血干细胞移植(hematopoietic stem cell transplantation,HSCT)是目前根治PNPD的唯一有效手段[8]。文献中,16例PNPD患者行HSCT,移植年龄为24个月(3.5~168个月)[3,5]。预处理方案包括清髓、降低强度,1例患儿未经预处理。大多数移植后的并发症与感染有关,其中2例死于移植后感染,2 例继发性移植物植入失败需要再次移植,总生存率为87.5%,所有14例存活者情况良好,中位随访18.3个月(8~108个月)。大多数患儿嵌合率为90%以上,仅2例尚需定期输注IVIG,大多数患儿在移植后都有一些运动、语音发育上的改善,提示及时、恰当的治疗对本病预后良好。利用慢病毒基因疗法治疗PNPD已经入尝试阶段[9]。
总之,当患者有反复感染,伴或不伴免疫性疾病、神经功能障碍、T细胞免疫缺陷、尿酸水平低时需考虑PNP基因变异致免疫缺陷病可能。基因序列分析可协助早期诊断,同时可对患者家族中携带者或高风险孕妇进行产前诊断,进而促进优生优育。对确诊患儿行HSCT,有助于提高此类患者的生存率及生活质量。
