高韧性三元层状陶瓷:从MAX相到MAB相
2021-05-21柏跃磊宋广平赫晓东齐欣欣郝兵兵张金泽
柏跃磊,尹 航,宋广平,赫晓东,2,齐欣欣,高 进,郝兵兵,张金泽
(1 哈尔滨工业大学 特种环境复合材料技术国防科技重点实验室/复合材料与结构研究所,哈尔滨 150080;2 深圳烯创先进材料研究院有限公司,广东 深圳 518000)
现代空天飞行器技术的发展对材料的耐热性能提出了越来越高的要求[1]。例如,高超声速飞行器在以5马赫以上速度飞行时,由于空气阻力而产生的超高温热流使其翼前缘、头锥以及推进系统部件所面临的温度高达2000 ℃以上。而新一代高推重比航空发动机涡轮前温度的提高则使涡轮叶片材料要承受越来越高的温度。传统的高温合金材料受限于其熔点已无法满足要求,而耐热性能更好的陶瓷则成为唯一选择。源自强共价和离子键合特征,陶瓷材料在表现出良好高温性能的同时也存在本征脆性的固有缺点,即强裂纹敏感性和低损伤容限[2]。这成为其实现工程应用的主要挑战,因此增韧成为陶瓷材料领域一个经久不衰的研究热点。
1996年,美国德雷塞尔大学的Barsoum教授等发现Ti3SiC2表现迥异于典型陶瓷材料的力学行为,即高断裂韧度、高损伤容限、可加工等[3]。这一开创性工作引发了世界范围内对三元层状化合物的研究热潮[4]。该类材料具有类似的纳米层状晶体结构(空间群P63/mmc),Barsoum教授将其命名为“Mn+1AXn”相,简称MAX相,其中M是过渡族金属元素,A是ⅢA或ⅣA族元素,X是C或N,n= 1~3[5]。MAX相化合物可以看作是NaCl型二元碳/氮化物晶格中插入一层主族原子后形成的。受MAX相的启发,在二元硼化物中插入单层或双层主族原子(典型如Al原子)也可形成被称为“MAB相”的三元层状过渡金属硼化物[6]。目前,该类材料已经成为近期陶瓷材料研究领域的研究热点[7]。虽然早在20世纪60年代大部分MAB相化合物就已经被发现[8-9],但直到2013年细致的结构和性能表征才陆续开展[10]。2015年,Ade等合成出一系列MAB相单晶并对其结构和硬度进行了研究[6]。而最近的实验工作显示MAB相(MoAlB[11]和Fe2AlB2[12])确实具有类似于MAX相的高断裂韧度和损伤容限以及良好的抗氧化性能,这对其潜在的结构应用极为重要。
三元层状陶瓷的非凡力学性能源自该类陶瓷的层状结构和弱化学键合[4],这就为陶瓷增韧提供了另一可行途径。进一步阐明该增韧机制不仅对筛选和理论设计高韧性层状陶瓷极为关键,同时也为理论预测陶瓷的宏观力学行为奠定基础。尤其是近期对取向MAX相陶瓷的研究进展更加凸显了从事这项研究的重大意义[13-15]。最近,键刚度模型的建立在实现对化学键强度的定量表征外[16],更重要的是揭示了三元层状陶瓷宏观断裂行为与微观化学键强度的关系。目前的结果显示,当最弱化学键刚度与最强化学键刚度的比值kmin/kmax大于1/2时,这类陶瓷材料表现出典型脆性陶瓷的低损伤容限和断裂行为,如三元碳化物(MC)nAl3C2和(MC)nAl4C3陶瓷[17-18];而当kmin/kmax小于1/2时,这类陶瓷则表现出高损伤容限和高断裂韧度,如MoAlB[19]和典型的MAX相陶瓷[20]。换句话说,三元层状陶瓷中最弱化学键强度要弱到小于最强化学键的1/2才能使其具有高断裂韧度和损伤容限。
作为过去20余年的研究热点,已经有大量的论文发表在国内外学术期刊上,涵盖制备工艺、力学行为、热力学性能、摩擦磨损行为、氧化行为等。及时总结在该领域的最新研究进展将有利于深入认识和理解三元层状化合物及其增韧机制,促进该类材料在更大范围内的工程应用。高断裂韧度和高损伤容限依然是三元层状陶瓷区别于传统陶瓷的本质特征,因此本文将重点探讨该类陶瓷的力学性能及其物理机制。本文主要包括以下两个部分:首先介绍MAX相的发展概况、晶体结构、纳米层板结构、高断裂韧度和损伤容限、键刚度模型及其在揭示MAX相非凡力学行为物理机制方面所起的作用。然后,将重点评述在三元层状硼化物MAB相领域的最新研究进展,具体包括MAB相的发展概况、纳米层状结构、制备工艺、力学性能及其晶体结构依赖性、Fe2AlB2在室温附近的磁热效应等。最后,将探讨三元层状陶瓷目前存在的问题和未来发展方向。
1 MAX相:一类具有非凡力学性能的三元层状碳化物和氮化物
1.1 发展历史与概况
早在20世纪60年代,奥地利维也纳大学物理化学研究所的Nowotny等就在实验中发现了后来被称为“MAX相”大部分三元层状碳化物和氮化物并报道了这些化合物的晶体结构和晶格常数[21-28]。随后在他们所撰写的综述中对这些发现进行了总结,报道了2种312型化合物(Ti3SiC2[25-26]和Ti3GeC2[27])和当时被称为“H相”的30余个211型MAX相化合物[29]。虽有零星报道,但是人们在之后的很长一段时间里几乎忽略了这一类化合物。这一时期最重要的发现是实验观察到了一种新型312型MAX相化合物Ti3AlC2[30]。
由于实验合成块体MAX相通常含有不同成分和含量的杂质,因此人们对其本征性能长期缺乏了解。Ti3SiC2是最早引起人们关注的MAX相化合物,早期得到了较多研究。早在1972年,Nickl等[31]就采用化学气相沉积(CVD)成功制备出Ti3SiC2单晶,而后续的表征显示它是一种很软的过渡金属碳化物。并且,它的硬度表现出明显的各向异性:垂直基面的硬度大约是平行该面的3倍。1987年,Goto和Hirai[32]证实了Nickl等[31]结果的正确性。相比之下,制备单相致密Ti3SiC2多晶陶瓷的道路更加曲折,因为在大部分情况下往往伴随着大量杂质相[33-35]。尽管如此,Pampuch等[34]也制备出了体积分数在80%~90%之间的多晶Ti3SiC2块体材料,显示其具有很高的刚度和良好的可加工性,杨氏模量和剪切模量分别为326 GPa和135 GPa。另外,他们还测出该材料的硬度为6 GPa,并指出刚度/硬度比更接近于韧性金属而被称为“韧性陶瓷”。另外,Iseki等[36]在1989年还报道了Ti3SiC2的热膨胀系数为9.2×10-6K-1。
MAX相领域的重大突破出现在1996年:美国德雷塞尔大学的Barsoum教授以Ti,SiC和石墨混合粉末为原料采用热压工艺在1600 ℃,40 MPa下保持4 h成功制备出几乎为单相的致密Ti3SiC2,特别是后续的实验表征显示了MAX相材料非同寻常的性能[3]。例如,不同于传统陶瓷材料,Ti3SiC2表现出金属和陶瓷的共同性能,如高损伤容限、高断裂韧度、低硬度、可加工、抗热震、高阻尼[37]、高刚度[38]、良好的导电导热性能[39]、抗氧化[40]等。后续的研究显示之前被人们长期忽视的211型MAX相化合物也具有类似性能[41-44]。这一非凡性能迅速引起了陶瓷研究界的广泛关注,形成了世界范围内持续至今的研究热点。研究的重点也逐渐从早期的Ti3SiC2转移到其他含铝MAX相化合物,如Ti2AlC和Ti3AlC2[45-47]。
人们认识另一大类413型MAX相化合物则要明显晚得多。1999年,Barsoum等发现文献中“Ti3AlN2”的c轴晶格常数(≈23 nm)明显高于相同结构的Ti3AlC2(≈1.8 nm),因此认为该“Ti3AlN2”并不是312型结构的[48]。后续的透射电镜(TEM)观察显示该化合物的真实结构实际上是Ti4AlN3[49],这也是当时唯一在实验中发现的413型MAX相化合物。然而,2006年后的实验进展改变了这一认识。首先,理论预测Ti4SiC3的成功促使人们把注意力转到了合成新MAX相中去[50-51]。但是,由于当时合成的新相(Ti4SiC3[51]和Ti4GeC3[52])均是以薄膜状态存在,人们普遍认为这些新型MAX相均是亚稳的,只能够在非平衡状态下形成(如外延生长的磁控溅射)。突破发生在Ta-Al-C体系中:2006年,几个课题组分别制备出Ta4AlC3块体材料[53-56],而对Ta4AlC3晶体结构的争论导致发现其存在两种晶体结构[55]。紧接着,Etzkorn等发现了V4AlC3[57],周延春等发现了Nb4AlC3[58]以及固溶体(V,Cr)4AlC3[59]。2009年,Etzkorn还合成出了单晶Ti4GaC3[60]。这些新的进展使人们意识到413型MAX相化合物的数量比当初预想的要多。
基于密度泛函理论(DFT)[61]的第一性原理方法在理解和预测MAX相的非凡性能方面扮演了重要角色[4],这成为MAX相研究的突出特点之一。其原因是合成单相三元层状陶瓷或者制备单晶材料比较困难,这对揭示其基本性能造成了不少的困难。另一方面,MAX相陶瓷是个很庞大的家族,其中包含近百种化合物,逐个实验研究它们的性能需要耗费大量的资源。早在1998年,Medvedeva等就采用DFT研究了Ti3SiC2的电子结构[62]。而第一性原理模拟解决了MAX相领域许多重要问题。例如,高的弹性刚度源自在M-C层片中M—C强共价键[63-65]。而相对较弱的M—A键导致了很多有趣的性能,例如室温下基面位错滑移和低剪切变形阻力[66-69]。限于篇幅,本文不能过多讨论这一问题,更多信息请参考相关文献[4]。
实验[5]和理论[73]结果均显示MAX相是间隙化合物,其中M原子之间的间隙位置由A和X原子填充(图1)。例如,由于211相沿c轴方向有4层M原子,因此如果不考虑晶格畸变的话晶格常数c应该是晶格常数a的4倍。也就是说,c/a比的理想值应该是4。以此类推,312和413相的c/a比分别应该为6和8。实际上,A和X原子插入晶格间隙将不可避免地引起晶格畸变,因此它们的c/a比会稍微偏离理想值。
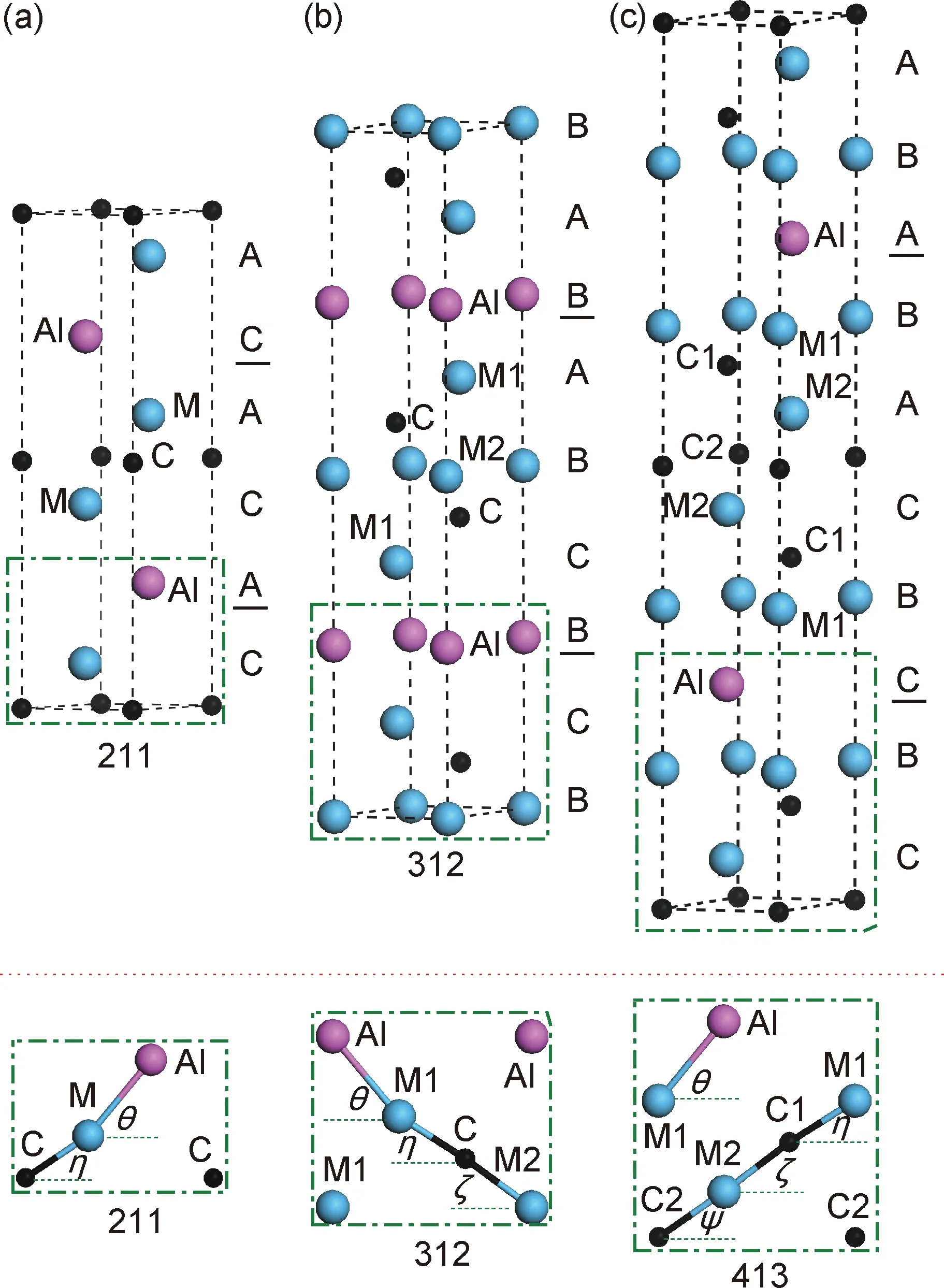
图1 MAX相的晶体结构图[20] (a)211;(b)312;(c)413Fig.1 Crystal structure of the MAX phases[20]
1.2 晶体结构与多态性现象
MAX相化合物都具有层状、六方的晶体结构,空间群为P63/mmc(194)[5]。当n=1时,称H相或211相,目前实验中已发现的MAX相绝大部分是H相,如Ti2AlC,Cr2AlC等;当n=2时,简称312相,代表性的化合物为Ti3SiC2;当n=3时,简称413相,其中Ti4AlN3最为大家所熟悉。虽然已有更高阶的MAX相化合物(n> 3)结构单元在透射电镜中被发现,但是没有合成出高纯块体材料。如图1所示,每个MAX相晶胞包括了2个化学式的原子,其结构可以描述为被一层A族原子插入的边共享M6X八面体,而该八面体的结构与其对应的二元化合物相同。不同数量的M-X层片数量是三种结构的主要区别。
312和413型MAX相均表现出有趣的多态性现象。图1实际上是MAX相最普遍也是最稳定的晶体结构,分别为M2AX,α-M3AX2和α-M4AX3。其他典型MAX相结构还包括β-M3AX2,β-M4AX3和γ-M4AX3。这些结构的内坐标如表1所示。到目前为止,Eklund等[74]将实验中所发现的MAX相多态性现象分为两种:一种是在312型(可能还有Ti4AlN3)结构中发现的,伴随着A层原子的剪切变形,其多态性表现为A组原子占据晶胞中不同的内坐标位置,同时过渡金属碳化物或氮化物单元保持不变[66,70,75-77];另一种只在Ta4AlC3中有报道,主要是M-X层片单元有所不同。Eklund等[74]认为前者是由剪切应变引起的,而后者最有可能是由热力学因素驱动的。

表1 MAX相的原子位置和内坐标
MAX相的多态性现象第一次引起人们的注意是在20世纪90年代末。当时随着对Ti3SiC2,Ti3GeC2以及Ti4AlN3研究的逐渐深入,人们发现使用不同方法例如Rietveld精修、高分辨透射电镜(HRTEM)等所确定的晶体结构存在差异[49,70-71,78]。当时人们认为这是一种伴随着Al/Si原子剪切变形的多态性相变,可能是在试样制备过程中产生的。实验中所观察到的两种Ti3SiC2结构分别被称为α-Ti3SiC2和β-Ti3SiC2。根据Farber等[70]的定义,α-Ti3SiC2和β-Ti3SiC2的差异在于Si原子占据了不同的2b Wyckoff位置:α-Ti3SiC2中Si原子的内坐标为(0, 0, 1/4),而在β-Ti3SiC2中为(2/3, 1/3, 1/4)。然而,这些都是建立在HRTEM照片之上,由于α-β相变似乎是在TEM制样过程中产生的,这使人们无法肯定其是否具有宏观代表性。直到2004年,Wang等[77]在采用同步辐射X射线衍射技术研究Ti3GeC2的压缩行为时发现,当压力升高到26 GPa时,α-Ti3GeC2转变为β-Ti3GeC2。另一方面,一项类似的研究[79]表明α-Ti3SiC2可以在小于61 GPa的静水压力下保持结构稳定性。然而在更高的静水压力下(≈90 GPa),则有α-β相变发生[80]。王京阳等[66]采用第一性原理方法从理论上揭示了Ti3SiC2在大剪切形下的多态性相变是通过Si原子在两个低能量处沿着基面(0001)滑移完成的。
第二种MAX相多态性已经在Ta4AlC3中得到实验证实。Manoun等[54]采用同步辐射X射线衍射技术研究了Ta4AlC3的压缩行为。他们将Ta4AlC3的结构设定为Ti4AlN3型,但是计算和实验获得的XRD图谱之间存在较大差别,他们将此归因于晶粒的择优取向。不久,林志军等[53]使用HRTEM发现他们所合成的热压Ta4AlC3表现出了与Ti4AlN3不同的晶体结构,而且同时进行的Rietveld精修证实他们所提出的结构与实验数据吻合得很好。非常有意思的是,Eklund等[55]的实验结果却表明他们所合成的Ta4AlC3具有和Ti4AlN3类似的晶体结构。另外,Etzkorn等[56]也几乎同时得到了相同的结果。基于以上的实验结果可以看出,Ta4AlC3存在两种晶体结构,被称作α-Ta4AlC3(与Ti4AlN3的结构相同)和β-Ta4AlC3。可以确定,Manoun等所研究的是β-Ta4AlC3。与312相的多态性不同,α-Ta4AlC3和β-Ta4AlC3的区别在于Ta2和C2原子占据不同的内坐标位置,而A原子位置保持不变。
γ-M4AX3则是在研究Ti4GaC3晶体结构时发现的:XRD精修[60]表明86.1%的Ga原子占据2c位置,其余的13.9%占据2d位置;过渡金属碳化物或氮化物层片保持不变。He等[72]认为这个新发现的Ti4GaC3存在两种晶体结构(多形体):一种是为广大研究者所熟知的α型结构;然而,第二种结构并不是大家熟知的β型结构,而是一种新的结构(γ型)。实际上,这种结构曾经在高分辨透射电镜实验的Ti4AlN3试样中观察到[49]。α-γ的相变能垒(≈0.01 eV/atom)比α-β(≈0.7 eV/atom)和β-γ(≈1.0 eV/atom)的低大约两个数量级(图2)。由此可见,α-γ相变应该比其他的多态性相变更容易发生,这也与实验中观察到的现象一致:用于透射电镜观察的减薄α-Ti4AlN3试样很容易转化为γ型[49]。而β相一旦形成,在动力学上应该非常稳定。这样的结果并不令人惊讶:从α到γ的转变仅需要A(Ga)原子从2c位置移动到2d位置。然而,α到β的转变则需要破坏强得多的M—X(Ti—C)共价键。类似的相变机理也适用于312相的α-β相变。

图2 Ti4GaC3多形体之间的相变能垒[72](TSa-b,TSa-c和TSb-c分别对应α-β,α-γ和β-γ相变的过渡态)Fig.2 Energy barrier among Ti4GaC3 polymorphs(TSa-b,TSa-c and TSb-c correspond to the transition states of α-β,α-γ and β-γ phase transitions, respectively)[72]
1.3 纳米层合板
MAX相沿c轴方向的堆栈次序是M,A和X原子层的交替排列(图1),其中M和A原子层之间是由较弱的M—A键连接,而M和X原子层之间由强M—X共价键连接[4]。也可以把MAX相看成是紧密连接在一起M—X单元层被弱化学键M—A连接。这一特有的晶体结构和化学键合特点使得MAX相在力学性能上表现出明显的各向异性和组织结构上的层状特征。值得指出的是,这一层状结构在晶体内部,尺度在纳米级别,因此MAX相也被称为“热力学稳定纳米层合板”[5]。
MAX相的纳米层状结构是其具有非凡力学性能的根本原因。这是由于MAX相中的弱结合(M—A键)在外力作用下极易萌生裂纹并扩展进而发生分层破坏(图3(c),(d))。这一层状结构对提高MAX相的断裂韧度极为关键,其机理为主裂纹在扩展过程中遇到MAX相晶粒中的弱结合面会发生偏转,从而使整个扩展过程中所需要的能量显著升高。实际上,该增韧机制与经典复合材料层合板和陶瓷基复合材料的增韧机制类似,只是存在尺度上的差异。此外,MAX相的基面位错在室温下是可动的[82],因此单晶MAX相晶粒在室温下即可以通过形成扭折带而发生微观塑性变形[81](图3(b))。这就意味着MAX相单层片之间存在着塑性层,其在外力作用下亦可通过塑性变形松弛裂纹尖端应力集中和耗散应变能从而进一步提高韧性和损伤容限。

图3 Ti2AlC的断口形貌[81] (a)断口全貌;(b)晶粒弯曲和扭折;(c)层间分层;(d)图(c)的放大图Fig.3 Fracture surfaces of Ti2AlC[81] (a)full view;(b)kink of grains;(c)delamination;(d)enlarged view of fig.(c)
1.4 高断裂韧度和损伤容限
MAX相化合物最令人惊奇的是其不同于传统脆性陶瓷材料的力学行为。该类材料显示出的高断裂韧度和高损伤容限是其区别于传统脆性材料的最主要特征。取决于具体的组织结构和测试方法,实验测定的MAX相化合物断裂韧度值在5~20 MPa·m1/2之间[83],显著高于传统陶瓷(1~3 MPa·m1/2),如Al2O3,Si3N4和SiC等[84]。与其他陶瓷类似,MAX相的断裂韧度随着晶粒尺寸的增大而增大。例如,Gilbert等发现细晶Ti3SiC2的断裂韧度值为8 MPa·m1/2,而相应的粗晶样品值为8.5~11 MPa·m1/2[85]。类似的现象也出现在Cr2AlC中:随着晶粒尺寸从2 μm增加到35 μm,Cr2AlC中的断裂韧度从4.7 MPa·m1/2提高到6.2 MPa·m1/2(图4(b))[86]。另外一个值得注意的现象是MAX相材料的断裂韧度表现出明显的各向异性。例如在晶粒高度择优取向的Nb4AlC3中,沿着[0001]方向加载时所测的断裂韧度为(17.9±5.16) MPa·m1/2,而垂直于该方向加载时断裂韧度降低到(11.49±1.38) MPa·m1/2[13]。此外,R曲线行为是MAX相材料裂纹扩展过程中表现出的另一重要特征。实验中发现细晶Ti3SiC2中裂纹开始扩展时的断裂韧度值为8 MPa·m1/2,而当裂纹扩展1.5 mm后断裂韧度增加到约9.5 MPa·m1/2(图4(a))。该行为在粗晶Ti3SiC2中表现得更加明显[87]。

图4 粗晶和细晶Ti3SiC2的裂纹扩展阻力随裂纹生长的演化曲线(a)[87]和晶粒尺寸对断裂韧度的影响(b)[83]Fig.4 Crack growth resistance[87](a) and effect of grain size on fracture toughness(b)[83] of fine and coarse Ti3SiC2
MAX相材料表现出非常高的损伤容限,通常采用维氏压痕测试后残余强度随压痕载荷的变化来表征。如图5(a)所示,在所研究的载荷范围内残余强度与载荷大小表现出明显弱的相关性,并且粗晶试样显示出更高的损伤容限[88]。这种高损伤容限与MAX相具有通过塑性变形将损伤范围限制在压痕周围的能力密切相关,同时也是无法在维氏压痕对角线处引发出显著裂纹扩展的原因。MAX相材料的这种高损伤容限还可以通过图5(d)直观感受到:经锤子反复敲击后的Ti2AlC表面只出现了几个凹痕而并没有沿压痕对角线处出现灾难性裂纹扩展,与金属材料中的实验结果类似[83]。

图5 MAX相的损伤容限 (a)Ti3SiC2,Ti3GeC2和Ti3Si0.5Ge0.5C2经维氏压痕测试后的四点弯曲强度[88];(b)46 μm Ti3GeC2试样的组织结构;(c)7 μm Ti3Si0.5Ge0.5C2试样的组织结构;(d)块体Ti2AlC经铁锤反复敲打后的表面形貌[83]Fig.5 Damage tolerance of the MAX phases (a)4-points bending strength of post-indented Ti3SiC2,Ti3GeC2 and Ti3Si0.5Ge0.5C2[88];(b)microstructures of Ti3GeC2 (46 μm);(c)microstructures of Ti3Si0.5Ge0.5C2 (7 μm);(d)surfaces of Ti2AlC block repeatedly hit with a steel hammer[83]
1.5 键刚度模型
材料的宏观性能是与其化学键合特征密切相关的,如强度、断裂韧度、弹性模量等。但是实验测定化学键强度极其困难,因此人们试图采用基于密度泛函理论的第一性原理方法从理论上计算化学键的强度。传统上,通过电子态密度和电荷密度分析可以定性分析同种化合物中化学键的相对强度。然而,这类常规方法无法实现对化学键强度的定量表征以及无法比较不同化合物中化学键的相对强度。针对这一问题,柏跃磊等将化学键在静水压力作用下的变形抗力即刚度(k)用于表征化学键强度,建立了一个称之为“键刚度”的理论模型[16,72]用于定量研究化学键强度k。该模型可简述如下:
d/d0=C0+C1P+C2P2
(1)
(2)
式中:k为化学键刚度,GPa;P为静水压力,GPa;d为某一静水压力下的键长,nm;d0为0 GPa时的键长,nm;Ci为最小二乘多项式拟合中的拟合系数(i=0,1,2)。
采用该模型可以很容易地计算出Ti4GaC3和含铝MAX相碳化物的化学键刚度(图6)。明显地,M—Al键在各体系中具有最低的化学键刚度,与电子结构的分析结果一致。其刚度值大约是对应M—C键刚度的1/3到1/2。对于M3AlC2和M4AlC3,与Al原子最近的M—C键具有最高的刚度,而在M4AlC3中其他两种M—C键则具有相近的刚度值。有趣的是,不同体系(M2AlC,M3AlC2和M4AlC3)中同一种化学键具有相近的刚度,并未受到结构中M-C层片数量的影响。

图6 M2AlC(a),M3AlC2(b)和M4AlC3(c)在0 GPa时的化学键刚度[20]Fig.6 Bond stiffness at 0 GPa in M2AlC (a), M3AlC2 (b) and M4AlC3 (c)[20]
化学键刚度与材料的宏观力学性能存在定量关系。若定义各化合物中的化学键刚度之和为“总化学键刚度”(total bond stiffness),图7显示了M2AlC,M3AlC2和M4AlC3的体积模量B和总化学键刚度之间存在正比例关系[20]。并且,仔细观察可以发现图中拟合直线的正比例系数S与所考虑化学键的个数n值(也等于化学式Mn+1AXn中的n)存在如下关系:

图7 M2AlC(a),M3AlC2(b)和M4AlC3(c)的体积模量和总化学键刚度的关系[20]Fig.7 Bulk modulus vs the total bond stiffness for M2AlC (a), M3AlC2 (b) and M4AlC3 (c)[20]
2SM2AlC≈3SM3AlC2≈4SM4AlC3≈nS≈0.256
(3)
(4)
则有
(5)
因此在普遍情况下,MAX相的体积模量决定于平均化学键刚度,二者之间存在简单的正比例关系。基于此,可以得出如下结论:(1)M—X键是决定MAX相体积模量的关键因素;(2)M—A键较低的化学键刚度是MAX相化合物比对应二元碳化物拥有较小体积模量的根本原因。
1.6 “足够”弱的层间结合:非凡力学性能的物理本源
键刚度模型还在揭示三元层状陶瓷非凡力学性能的物理机制方面起到了关键作用(表2)[4]。众所周知,MAX相晶格中弱M—A键和纳米层状结构是其表现出非凡力学性能的根本原因[5]。然而,MAX相化合物Ti2SC却表现出类似于传统陶瓷的脆性断裂行为,即在维氏压痕对角线处出现主裂纹扩展(图8(a))[95]。键刚度计算结果表明Ti2SC中的Ti—S键刚度为578 GPa,而Ti—C键刚度为752 GPa。换句话说,最弱的Ti—S键依然弱于Ti—C键,但是最弱和最强化学键的刚度比值kmin/kmax为0.78。而对于除Ti2SC的绝大部分MAX相材料,这一比值在1/2到1/3之间。因此,可以将kmin/kmax作为三元层状陶瓷宏观力学行为的理论判据:当kmin/kmax大于1/2时,这类陶瓷材料表现出典型脆性陶瓷的低损伤容限和断裂行为,如Ti2SC(图8(a));而当kmin/kmax小于或接近1/2时,这类陶瓷则表现出高损伤容限和高断裂韧度,如Ti2AlC(图8(b))。维氏压痕实验中发现,前者出现沿对角线方向的裂纹形核与扩展,而后者则始终没有该现象出现,表现出极高的损伤容限。这一发现表明MAX相具有非凡力学性能,不仅要求M—A键要弱于M—C键,而且还必须“足够”弱。

图8 MAX相的维氏压痕与最弱和最强化学键的相应刚度比值kmin/kmax (a)Ti2SC[95];(b)Ti2AlC[107]Fig.8 Vickers’ indentation and corresponding ratio of the bond stiffness of the weakest bond to that of the strongest bond kmin/kmax of the MAX phases (a)Ti2SC[95];(b)Ti2AlC[107]

图9 (HfC)nAl3C2和(HfC)nAl4C3的晶体结构[17]Fig.9 Crystal structure of (HfC)nAl3C2 and (HfC)nAl4C3[17]
实际上,M—A键强度决定了MAX相中纳米层片之间的结合强度。当裂纹在MAX相晶粒内部扩展遇到这个弱结合面时,如果这个结合面较强裂纹会直接穿过该面呈直线扩展,而只有当该结合面足够弱时裂纹会在此处改变扩展方向沿该弱结合面扩展,即发生裂纹偏转。显然,后者才会显著增加裂纹扩展长度、提高材料的断裂韧度。实际上,当MAX相晶粒由随机排布变成取向排布可强化此效应而进一步提高断裂韧度,例如常规多晶Nb4AlC3的断裂韧度为(7.1±0.3)MPa·m1/2[98],而当晶粒取向排列后增加至(17.9±5.16) MPa·m1/2[13]。类似的增韧机制也出现于典型的陶瓷基复合研究中:只有纤维和基体之间的界面结合强度适度弱时,纤维才能起到增韧作用[108]。基于以上所述机理,kmin/kmax可以用于表征MAX相内部不同方向的相对亚界面结合强度和不同方向的裂纹扩展阻力,因而可以成为三元层状陶瓷宏观力学行为的理论判据。
这一判据不仅对MAX相化合物有效,也适用于其他三元层状化合物。(MC)nAl3C2和(MC)nAl4C3(M=Zr或Hf,n= 1, 2, 3...)[109]是另一类三元层状化合物,前者的空间群为P63/mmc,而后者的是R3m。对该系列Hf系[17]和Zr系[18]化合物的理论计算结果表明其最弱结合面在Al-C结构单元的中间位置,最弱和最强化学键的比值kmin/kmax均大于1/2。而Zr-Al-C系的实验结果表明在这些化合物维氏压痕对角线附近出现明显的主裂纹扩展[101-103],显示了该类化合物的低损伤容限和断裂韧度。需要指出的是,三元层状陶瓷的增韧机制源自层状晶体结构和弱层间结合,而与所研究化合物的组成元素数量没有关系,因此该理论判据同样适用于具有类似层状晶体结构和化学键合的其他化合物。
2 MAB相:具有高断裂韧度和损伤容限的三元层状过渡金属硼化物
自1996年以来,Barsoum等[7]的开创性工作使人们认识到MAX相材料具有较好的综合性能,如良好的导热导电性能、较低的维氏硬度和剪切模量、高杨氏模量、高熔点、优异的耐热冲击性能、可加工性、较高的屈服强度、高温塑性、高热稳定性和良好的抗氧化性能。正是这些优良综合性能激发了人们对三元层状过渡金属硼化物MAB相陶瓷的研究兴趣,由于二者极为相似的晶体结构与物相组成,人们相信在MAB相材料中应当具有与MAX相材料相似的优良性能。
2.1 二元硼化物:一类具有广阔应用前景的超高温陶瓷
二元硼化物由于具有高硬度、高温抗蠕变性、高耐磨性、高熔点、耐腐蚀、良好的导热与导电性等性能[110]而在保护电镀涂层、集成电路、高温耐火材料、切削刀具、电子元件、耐磨器具等方面[111]受到高度重视。
二元硼化物能够具有众多优异性能的原因是化学键的成键表现出多样性。大多数硼化物具有B—B共价键、M—B离子键、M—M金属键,这决定了其高熔点、高硬度、耐腐蚀等特性[111]。以典型硼化物陶瓷ZrB2和TiB2为例,二者具有相似的晶体结构,内部硼原子层与金属原子层交替出现,形成的B—B共价键和M—B离子键使位错难以移动,决定了其高硬度、高熔点性能,外部离域π键电子决定了其良好的导电与传热性能[112]。典型二元硼化物性能见表3[113]。

表3 典型二元硼化物材料性能[113]
虽然二元硼化物具有较好的高温性能,但由于其烧结温度高、低断裂韧度(KⅠC一般小于4 MPa·m1/2)和较差的抗热震性能等缺点阻碍了材料的进一步应用[114]。虽然有学者通过加入第二相、相变增韧等方法不断提高材料的断裂韧度和其他性能,但这些方法普遍存在制备工艺复杂、生产成本高等问题。
2.2 从原子尺度增韧:引入Al原子层形成具有弱结合面的三元层状硼化物MAB相
MAX相的高断裂韧度和损伤容限提供了改善二元硼化物本征脆性的另一方法,即在二元硼化物晶格内部引入一层主族原子形成弱结合面。而2016年后,MoAlB[11],Fe2AlB2[12]等典型MAB相块体制备工艺的突破和后续的性能表征验证了这种思路的可行性。
实际上,Ade等在2015年就提出了“MAB相”的概念[6]:他们在研究了一系列三元层状硼化物,包括Cr2AlB2,Cr3AlB4,Cr4AlB6,MoAlB以及WAlB,Mn2AlB2和Fe2AlB2后,将这一类由M-B层之间插入不同数量金属层形成的硼化物,参照MAX相的命名方式将其称为“MAB相”。这里的M代表过渡金属元素,A代表ⅢA、ⅥA族元素,B代表元素硼。Ade等最初用通式(MB)2Aly(MB2)z(y=1,2,3,…;z=0,1,2,…)表示该系列化合物的化学式。但是,2019年Cr4AlB4型MAB相结构的发现显示该通式不再适用,而只能将其修改为(MB)2xAly(MB2)z(x=1,2,…;y=1,2,3,…;z=0,1,2,…)。目前已发现的MAB相主要分为以下类型:MAB型(空间群Cmcm)[119-120],M2AB2(空间群Cmmm)[8-9],M3AB4(空间群Immm)[120],M4AB6(空间群Cmmm)[6],M4AB4型(空间群Immm)[106, 121]和Ru2ZnB2型(空间群I41/amd)[122]。
2.3 MAB相的多尺度层状结构
图10是部分典型MAB相化合物的晶体结构。除“111”型MoAlB结构中含有2层A(Al)原子外,其他MAB相结构中仅有1层A(Al)原子。晶体结构中的M-B单元被A(Al)原子层分割而具有明显的微观层状结构特征(图11)。含有1层A(Al)原子时,MAB相中存在的化学键主要是强共价键M—B键和B—B键、弱化学键M—A和A—B键(表2)[105]。换句话说,A(Al)原子与M原子的结合处于弱结合状态。这使得MAB相晶体易在M层与A(Al)原子层之间发生解理断裂。而对于含有2层A(Al)原子的“111”型MAB相结构,情况则有所不同:最弱结合面出现在相邻的两层A(Al)原子之间[19]。

图10 MAB相的晶体结构Fig.10 Crystal structure of the MAB phases

图11 MAB相的透射电镜照片 (a) Fe2AlB2单晶[101]方向扫描透射电镜图[115];(b)图(a)相应的能谱图[115];(c)沿[100]和[001]方向带有倾斜边界缺陷的Cr2AlB2晶粒透射电镜图像[118];(d)NaOH处理后MoAlB晶体透射电镜图[116];(e)Cr3AlB4晶粒透射电镜图像,白色箭头为堆垛层错[117]Fig.11 TEM images of the MAB phases (a)HAADF-STEM of Fe2AlB2 single crystal [101][115];(b)corresponding EDS map of fig.(a)[115];(c)HAADF-STEM image of Cr2AlB2 grain with tilt boundary defect with domains along [100] and [001][118];(d)TEM image of MoAlB crystal after NaOH treatment[116];(e)TEM image of Cr3AlB4 grains, stacking faults as indicated by white arrows[117]
微观尺度上的层状结构和弱层间结合导致了MAB相在受载破坏时表现出明显的分层破坏特征(图12(d),(e))[123],这与得到广泛研究的MAX相化合物类似。其机理是当扩展着的裂纹遇到弱结合面后扩展方向发生偏转形成层间开裂。这种破坏所产生的层片厚度在数百纳米范围,因此这种较大尺度上的层片厚度处于亚微米尺度。分层破坏有效延长了裂纹扩展路径和增大了裂纹扩展阻力,避免了灾难性断裂的发生,提高了MAB相在整个服役过程中的安全性和可靠性,这是包括MAB相的三元层状陶瓷具有较高断裂韧度和损伤容限的根本原因之一。

图12 MAB相的多尺度层状结构 (a)MoAlB透射电镜图像[11];(b)MoAlB二次电子SEM图像[11];(c)MoAlB晶粒的高倍SEM照片[11];(d)Cr2AlB2晶粒分层[123];(e)Fe2AlB2晶粒分层[123]Fig.12 Multi-scale layered structure of the MAB phases (a)TEM micrographs of MoAlB[11];(b)secondary electron SEM micrograph of MoAlB[11];(c)high magnification SEM image of MoAlB grains[11];(d)layered grains of Cr2AlB2[123];(e)layered grains of Fe2AlB2[123]
2.4 块体MAB相的合成和基本力学性能
合成制备工艺的突破是对MAB相化合物进行结构和性能表征以揭示其非凡行为的前提。早在1942年,MoAlB就已经被Halla和Thurry发现[124],而Jeitschko[119]则在1966年确定了其正确的晶体结构。但是,早期人们只获得了一系列MAB相化合物的单晶,因此只能对其部分性能进行表征,例如晶体结构、显微硬度、电阻率等[6]。由于具有良好的磁致冷性能[10]和较低的原材料成本,单晶合成的重点主要放在了具有优异磁热性能的Fe2AlB2上。2011年,Elmassalami等[125]以Fe,Al,B三种单质粉末为原料,首次通过电弧熔炼法成功制备了Fe2AlB2单晶,主要杂质为Fe4Al13。2013年,Tan等[10]按照原料摩尔比Fe∶Al∶B=2∶3∶2,经电弧熔炼后用盐酸处理,再掺入过量Ga在900 ℃的真空环境下煅烧7天,最后离心去除金属Ga,成功制备了高纯度Fe2AlB2单晶。2015年,Du等[126]采用电弧熔炼结合熔体纺丝方法,以按照化学计量比混合的单质粉末为原料制备出高纯Fe2AlB2单晶,大大减少了原料粉的消耗。而同样在2015年,Ade等[6]在Al熔体中以单质粉末为原料生长出一系列MAB相化合物单晶,包括MoAlB,Fe2AlB2,Mn2AlB2,WAlB,Cr2AlB2,Cr3AlB4,Cr4AlB6单晶(图13)。

图13 MAB相单晶形貌[6]
早期所制备的单晶MAB相样品尺寸通常很小(图13),因此无法对其进行全面的性能表征。对MAB相更为全面的认识需要大块体MAB相制备工艺的突破。2016年,美国德雷塞尔大学的Barsoum教授[11]课题组以MoB与Al为原料,采用反应热压工艺在1200 ℃,39 MPa条件下保持348 min成功制备出相对密度为(94±1)%的MoAlB块体,发现了该材料表现出高损伤容限,掀起了MAB相的研究热潮。截至目前,已经成功制备出的块体MAB相材料主要为MoAlB,Fe2AlB2和Mn2AlB2,所用的制备工艺主要是采用热压(HP)和放电等离子烧结(SPS)方法在1050~1600 ℃,30~60 MPa的压力下保温10 min至近348 min不等(表4)。合成块体MAB相的原料主要为二元硼化物和单质Al,但是也有例外:2017年,柏跃磊课题组[12]采用元素粉末(Fe,Al和B)在1200 ℃保温30 min首次制备出“212”型MAB相化合物Fe2AlB2,显著降低了合成时间。值得注意的是,目前所合成的MAB相材料均含有少量杂质,主要包括Al2O3、二元硼化物和金属间化合物。前者主要是所用的原料Al粉颗粒表面氧化以及制备环境气氛中含有残余氧气所造成的,而后两者则主要归因于原料之间的反应不够完全。

表4 部分典型MAB相材料的制备工艺
制备工艺的突破为全面表征MAB相的力学性能提供了条件。表5是MoAlB,Fe2AlB2和Mn2AlB2的基本力学性能。MAB相材料的维氏硬度在10 GPa左右,明显高于MAX相材料(3~8 GPa)[83]。与之类似,接近2000 MPa的压缩强度也显著高于MAX相(380~1750 MPa)[83]。然而,弯曲强度在230~450 MPa之间,明显低于其压缩强度而与MAX相(200~600 MPa)材料相当[83]。因此,MAB相材料的压缩强度和弯曲强度的比值在3~8之间,高于MAX相材料(2~3)。需要指出的是,典型陶瓷材料由于低延性和相对较低的裂纹扩展阻力,该比值接近于10[133];而金属材料通常在发生很大的塑性变形后裂纹才开始扩展,具有很高的裂纹扩展阻力,因此二者差别不大。由此可见,从力学性能角度来看三元层状陶瓷介于典型的陶瓷材料和金属材料之间。并且与MAX相相比,MAB相的性能更加接近于典型的陶瓷材料。这也可以从它们的断裂韧度看出:不同课题组测定的断裂韧度值在4.3~5.4 MPa·m1/2范围内,稍低于MAX相材料(4.6~7.8 MPa·m1/2)[83]。

表5 部分典型MAB相块体的力学性能
众所周知,金属材料具有良好的韧性和损伤容限,但是受熔点的限制其使用温度无法进一步提高,目前耐热性能最好的高温合金使用温度也不超过1150 ℃[137]。陶瓷的耐热性良好但其本征脆性是其实现工程应用的主要障碍,若一步到位直接将其应用于工程将带来许多难以解决的问题。由于具有共价键、离子键和金属键的混合化学键合特征[63],三元层状陶瓷处于典型金属向典型陶瓷的过渡区域,性能也介于二者之间,有效弥合了金属和陶瓷之间的性能鸿沟[138]。通过材料筛选和工艺控制,三元层状陶瓷可实现耐热性和韧性的平衡统一,满足实际工程需求。
2.5 损伤容限和断裂韧度的结构依赖性:实验结果和理论考虑
包括MAB相的三元层状陶瓷最吸引人们注意的是其损伤容限和断裂韧度。美国德雷塞尔大学的Barsoum课题组首先发现了在MoAlB维氏压痕的对角线处没有出现主裂纹扩展(图14(a))[11],而是表现出类似于金属的损伤行为,表现出高损伤容限。随后,柏跃磊课题组也在Fe2AlB2中发现了类似的行为(图14(b))。同时,他们采用单边切口梁(SENB)法测定了MAB相(Fe2AlB2)的断裂韧度((5.4±0.2) MPa·m1/2)[12],确认了该类材料相对典型陶瓷的高断裂韧度。此外,MAB相的断裂过程也表现出非灾难性的破坏行为,即在到达最大应力时并未突然降低,而是呈阶梯状卸载(图15)。但仔细观察可以发现,MoAlB和Fe2AlB2的维氏压痕损伤特征也存在明显区别(图14):前者的压痕周围没有发现裂纹[11],后者却出现了明显的裂纹萌生和扩展,但是并没有主裂纹产生,并且裂纹在扩展过程中发生了明显的桥连和偏转[12]。需要指出的是,MoAlB和Fe2AlB2具有不同的晶体结构,最重要的区别是前者含有2层Al原子,而后者只有1层(图10)。

图14 MoAlB(a)[11]和Fe2AlB2(b)[12]的维氏压痕Fig.14 Vickers’s indentation of MoAlB (a)[11] and Fe2AlB2 (b)[12]

图15 采用SENB法测定Fe2AlB2断裂韧度的载荷-位移曲线[12]Fig.15 Plot of load vs displacement recorded during SENB bending test for fracture toughness of Fe2AlB2[12]
采用基于第一性原理的“键刚度”模型分析表明,MoAlB中最弱化学键是相邻2层Al原子层之间所形成的Al—Al键,这与典型MAX相材料不同。尤其值得注意的是,MoAlB中Al—Al键显著弱于Mo—Al键且前者的刚度与最强B—B键刚度的比值小于1/2(表2),因而弱Al—Al键是MoAlB相具有高断裂韧度和损伤容限的根本原因。实验上也证实了MoAlB具有和典型MAX相类似的损伤行为(图14(a))。如前所述,与MAX相不同,不同成分的MAB相化合物具有不同的晶体结构,如MoAlB型,Cr2AlB2型,Cr3AlB4型,Cr4AlB6型等(图10)。这显然会导致该类化合物表现出相对复杂的性能和行为,同时也为调控该类材料的性能提供了条件。采用“键刚度”模型对Cr-Al-B系MAB相化合物的理论分析[105]表明,晶体结构对MAB相陶瓷的宏观力学行为具有显著影响:在含有2层Al原子的CrAlB中,最弱化学键是Al—Al键,而Al—B(1)键则是含有1层Al原子的Cr2AlB2,Cr3AlB4和Cr4AlB6的最弱化学键(表2)。尤其需要注意的是,CrAlB的kmin/kmax小于Cr2AlB2,Cr3AlB4和Cr4AlB6的相应值,即含有2层Al原子的MAlB型较含1层Al原子的(MB)2Al(MB2)z(z=0, 1, 2)型具有更高的损伤容限和断裂韧度。需要指出的是,CrAlB与MoAlB的结构相同,而Cr2AlB2与Fe2AlB2的结构相同。
Cr2AlB2,Cr3AlB4,Cr4AlB4和Cr4AlB6的弱结合面存在于Al原子层和近邻的Cr原子层之间,表征其结合强度的化学键包括了Cr(1)—Al和Al—B(1)键(图10)。因此,虽然Cr2AlB2的kmin/kmax小于1/2,但其Cr(1)—Al键与最强化学键的比值却大于1/2,若取二者平均值则该比值依然大于1/2。而Cr4AlB4则是新近在实验中发现的一种MAB相化合物[121],晶格中也仅含有1层Al原子。然而,键刚度分析[106]表明其力学行为不同于其他含有1层Al原子的MAB相:其最弱的Al1—B键刚度(574 GPa)与最强的Cr—B键刚度(1190 GPa)比值为0.48。即使考虑到Cr1—Al键的贡献,平均kmin/kmax值依然在0.5附近。因此可以预测出Cr4AlB4具有类似于MoAlB和典型MAX相化合物的高损伤容限。该化合物含有的Cr,Al元素氧化后均可以形成致密的氧化膜而可能具有良好的抗氧化性能,因此Cr4AlB4在高温领域具有潜在应用前景,急需未来进一步的实验和理论研究以揭示其本征性能。
2.6 Fe2AlB2室温附近的磁热效应:一种有前景的磁致冷材料
磁热效应(MCE)是指在绝热条件下磁性材料受外界磁场作用而产生温度变化的现象。它于1881年被Warburg[139]发现,现在已经被广泛应用在磁制冷技术中。等温磁熵变(ΔSm)、绝热温度改变(ΔTad)和相对制冷能力(RCP)是衡量磁热效应的重要参量。Fe2AlB2具有较高的等温磁熵变ΔSm、绝热温度改变ΔTad和相对制冷能力RCP,尤其是其铁磁-顺磁转变温度(居里温度Tc)在室温附近,原材料丰富、来源广泛且成本低廉,在磁制冷应用方面具有巨大潜力。
金属硼化物通常被认为是硬磁材料,比如世界上最强的永磁材料Nd2Fe14B[141],其磁硬度源自稀土元素亚晶格的强各向异性。虽然大部分过渡金属硼化物为软磁体,但是一般磁性二元硼化物(如FeB和MnB)因其居里温度较高(598 K[142]与562 K[143]),并不符合磁制冷材料的要求。2013年,Tan等[10]成功制备出Fe2AlB2单晶和粉末,并测试了其ΔSm,ΔTad,RCP,Tc等磁热效应参数(表6)。他们发现Fe2AlB2的居里温度在室温附近,表明了其在磁制冷领域具有巨大潜力。因此,Fe2AlB2成为目前MAB相领域的研究热点,受到广泛关注。2015年,Du等[144]测试了熔体纺丝法制备Fe2AlB2的磁热性能,并在制备原料中添加Mn元素,降低了Fe2AlB2的居里温度。虽然ΔSm,ΔTad和RCP略有减小,但在ΔSm-T曲线中出现了一段直线,这显示通过调控Fe2AlB2成分可以使其在更接近常温的较宽温度范围内得到应用。随后,其他研究者通过添加Ga,Ge[145],Co[146],Cr,Ni[123],Al[147]等元素来调节Fe2AlB2的磁热反应,这为优化Fe2AlB2磁热性能提供了可行思路。

表6 Fe2AlB2和常见磁制冷材料磁热特性参数[10,140,149-151]
2017年,柏跃磊课题组采用原位反应热压法实现了Fe2AlB2的快速大批量制备[12],而且Fe2AlB2具有优异的抗热震性能[135]。这意味着与其他磁制冷材料相比,Fe2AlB2在使用时将具有更好的热稳定性。Zhang等[148]的研究表明,在0.5 T的磁场中,经过磁制冷环境(active magnetic regeneration,AMR)下500000周次的循环后,Fe2AlB2样品的磁热性能与微观结构都能保持不变,表明了其非凡的磁制冷稳定性。最近,柏跃磊课题组[140]通过DFT模拟与实验测试发现,Fe2AlB2磁化强度受制备工艺与产物形态影响不大,但由于Al2O3,Fe-Al间化合物等杂质的存在,ΔSm和RCP值高于单晶值,但ΔTad值低于单晶的结果。为了推广Fe2AlB2应用,提高产品纯度将是下一步改进的重要方向(图16)。

图16 反应热压合成Fe2AlB2的磁性能[140] (a)磁矩随温度的变化曲线(施加磁场为5 mT),顶部插图是2 K时的磁滞回线,底部插图是磁矩随温度曲线的一阶导数;(b)265~325 K温度区间的等温M-H曲线;(c)接近居里温度TC的Arrott曲线族;(d)在2~5 T区间内的磁熵变Fig.16 Magnetic properties of Fe2AlB2 fabricated by hot pressing[140] (a)temperature dependence of magnetic moment with an applied magnetic field of 5 mT from 2 K to 350 K. Top inset: field-dependent magnetization (magnetic hystersis loop) measured at 2 K. Bottom inset: derivative for temperature dependence of magnetic moment;(b)isothermal M-H curves from 265 K to 325 K measured in fields of up to 5 T;(c)arrott plots obtained near TC;(d)magnetic entropy changes calculated for magnetic field changes of 2-5 T
3 结束语
三元层状化合物以其兼具金属和陶瓷的共同特性成为结构陶瓷领域20余年的研究热点。高损伤容限和高断裂韧度是该类材料区别于传统陶瓷材料的根本特征。“键刚度”模型的建立不仅实现了对化学键强度的定量表征,而且从理论揭示了纳米层片之间的弱结合在三元层状陶瓷增韧机制中所扮演的核心角色。目前,经过来自不同国家和地区众多学者的长期探索,人们对MAX相化合物的结构和性能有了较为全面的认识。除继续开发具有优异工程性能的MAX相化合物,针对具体的应用场景进行应用基础研究和应用研究显得更为关键。MAX相拥有的独一无二性能——易加工、低摩擦因数、热和结构稳定以及良好的导电导热性能——使其在以下领域具有应用潜力:旋转电气触点和轴承、加热元件、喷嘴、换热器和加压模具。其中许多应用目前正在进行实地测试,并处于不同的开发阶段。例如,针对MAX相在航空发动机关键结构上的应用基础研究工作已经得到了广泛开展,虽然这通常需要较长的时间(20~30年)和大量的人力物力投入。目前MAX相实现工程应用的第一个障碍是与其他结构陶瓷(如SiC和氧化铝)相比,粉末和烧结组分的成本相对较高。另一个障碍是这些固体的新颖性使得一些研究人员不愿意使用它们替代已经使用了多年而更熟悉的固体。最后,缺乏无压烧结粉末也减缓了这些材料的应用和进一步发展。因此,尽快研究解决这些问题是目前MAX相领域研究的未来方向。
与MAX相相比,MAB相的研究则仍然处于非常初级的发展阶段。本文只是简单介绍了该类材料明显区别相应二元硼化物的结构和性能特点。MAB相具有高强度、导电导热等优异性能,尤其在1050~1200 ℃的温度区间采用常规的热压和放电等离子工艺即可制备出致密的块体材料,这与二元硼化物明显不同。结构和成分的多样性一方面增加了认识MAB相的难度和复杂度,另一方面导致了性能的多样性,但同时也丰富了调控和设计该类材料以实现工程应用的手段。虽然很多MAB相化合物早在20世纪40年代就已经被发现,但是2016年后学术界才真正注意到这类材料,目前对其结构和性能的认识极为有限。因此,合成和表征现有已知MAB相化合物的结构、力学性能、物理性能以及基于应用背景的使役行为是现阶段的重要任务之一。
目前已经在实验中发现的三元硼化物已经有60余种,而MAB相家族则会随着研究的开展变得更加庞大。除了M,A或者B位置的固溶体外,A组原子层数、M-B单元厚度和B-B链的方向等可以作为设计新型MAB相化合物的自由度。利用类似的方式,MAX相化合物的数量从2000年左右的50种增加到现在的150余种[152]。没有理由相信MAB相化合物的数量不会随着研究的不断推进而增加。
在发现和设计新型MAB相化合物的过程中,基于密度泛函理论(DFT)的第一性原理数值模拟可以扮演重要角色,正如其在理解MAX相化合物的非凡性能和发现新型化合物时所起的重要作用一样[4]。例如,DFT模拟首先揭示了弱M—A键是MAX相具有高断裂韧度和损伤容限的根本原因,而“键刚度”理论模型的建立则进一步表明只有相对M—X键“足够”弱的M—A键才能产生这种非凡力学行为。这一机制也同样适用于MAB相化合物:弱Al—Al键是MoAlB表现出高损伤容限的原因,并且含有2层Al原子的MAB相化合物较含有1层Al原子的具有更高的损伤容限和断裂韧度,令人兴奋的是这一结果与实验一致。DFT模拟在预测Cr-Al-B系MAB相化合物的稳定性[105]和热物理性能[140]方面也有良好表现。这些成功足以证明DFT模拟在发现新型MAB相化合物和预测其性能方面的有效性。为了进一步深入理解众多MAB相化合物的结构和性能,采用基于密度泛函理论的第一性原理方法理论研究该类材料以实现具有潜在工程应用前景材料的快算筛选及理论设计显得极为迫切。
由于已经展现出良好的应用前景,MoAlB和Fe2AlB2是目前研究较多的MAB相化合物。MoAlB具有良好的抗氧化性能、弱温度相关的热导率和弹性模量以及极为优良的抗压强度。室温时,与其他工程陶瓷相比MoAlB具有良好的韧性和抗热震性能。理解力学性能、抗氧化和其他腐蚀环境的性能随杂质、固溶和组织结构的演化将会有助于确定它的局限和潜在应用领域。事实上,MoAlB拥有了二元硼化物MoB的众多优点(例如高/热电导率和强度)和明显更加优越的抗氧化性能,并且可以在低至1200 ℃时成功制备。在应用方面,较高的强度和硬度使其可能作为抗冲击和摩擦涂层使用,而最近的摩擦磨损实验结果[153]则对该应用提供了部分支撑。
Fe2AlB2的居里温度接近于室温,因此其伴随着铁磁-顺磁转变的磁热效应(MCE)出现在室温附近。考虑到较高的等温磁熵变、绝热温度改变和相对制冷能力,Fe2AlB2在磁致冷领域具有应用前景,因此目前对Fe2AlB2的研究最为活跃。受益于制备工艺的突破,人们对其基本力学和物理性能的认识逐渐变得清晰和深入。与其他磁致冷材料相比,Fe2AlB2不含有稀土和有毒元素,原材料来源广泛且价格低廉,相对较小的热容和较高的热导率[140],这些使其工程应用具有明显优势。但是目前依然有很多问题有待解决,因此对Fe2AlB2的后续研究依然是必要的。例如,真实状态下的磁致冷通常是循环进行的,而目前的研究都是针对准静态条件进行的。因此Fe2AlB2的MCE随着磁饱和循环频率如何变化尚待厘清。此外,还要通过控制化学成分、固溶、组织结构以及复合化进一步提高Fe2AlB2的磁致冷性能。而这里,基于DFT模拟的理论设计可以加快材料的设计和筛选。
最后,包括MAX和MAB相的三元层状化合物实际上已经存在了70余年。最近20余年的研究显示了该类材料不同于传统陶瓷和金属的非凡性能,使其在结构和功能场合具有潜在应用前景。限于篇幅,本文无法详尽综述本领域的所有信息。希望本文可以刻画出该领域研究的整体框架和脉络,更重要的是鼓励对这类有趣材料进行更加深入的基础和应用研究,以阐明许多悬而未决的科学与技术问题,尽快实现该类材料的工程应用。
