miR-194通过调控KLF15表达降低缺氧诱导的心肌细胞损伤
2021-05-21韩芬杨晓
韩芬 杨晓
(1郑州铁路职业技术学院药学院基础医学教研室,河南 郑州 450000;2河南中医药大学第一附属医院耳鼻喉科)
急性心肌梗死严重威胁着人类健康及生活质量,而心肌组织缺血缺氧导致的心肌细胞坏死凋亡是重要原因〔1〕。缺氧在多种肿瘤细胞增殖与凋亡、新生血管形成、侵袭与转移等生物学行为的改变中发挥重要作用,miRNA是一类长度为19~24 nt的非编码RNA,调控其靶mRNA的翻译和表达可以改变缺氧诱导肿瘤生物学行为〔2〕。研究发现miR-195通过下调c-myb增强缺氧/复氧损伤诱导的心肌细胞凋亡〔3〕;过表达miR-29b1可抑制缺氧诱导的心肌细胞凋亡〔4〕。而miR-194可抑制喉鳞状细胞癌增殖〔5〕,miR-194通过调节鼠双微粒体基因(MDM)-2表达调节卵巢癌细胞中的紫杉醇抗性〔6〕,但miR-194对缺氧的心肌细胞的影响还尚未见报道。KLFs属于锌指转录因子亚家族,参与早期的胚胎生长发育、细胞分化、组织器官的形成、原癌基因的突变和血管生成等生理病理过程,KLF15是KLF家族的重要成员,具有同样的功能〔7〕。而KLF15在缺氧诱导的心肌细胞凋亡中作用尚不清楚,且miR-194与KLF15之间的调控关系也不清楚,本研究旨在探讨miR-194对缺氧诱导的心肌细胞损伤的影响及其可能作用的分子机制是否与KLF15有关,为心肌细胞损伤及其导致的相关疾病治疗提供新靶点和新方向。
1 材料和方法
1.1材料 心肌细胞H9C2购自中国科学院上海细胞库;胎牛血清、胰蛋白酶、DMEM培养液购自Gibco公司;兔抗人KLF15多克隆抗体、兔抗人CyclinD1多克隆抗体、兔抗人酶切半胱氨酸天冬氨酸蛋白酶(caspase)-3多克隆抗体、兔抗人p-STAT3多克隆抗体、兔抗人STAT3多克隆抗体、兔抗人β-actin多克隆抗体、辣根过氧化物酶(HRP)标记的山羊抗兔IgG均购自上海煊翎生物科技有限公司;二喹啉甲酸(BCA)试剂盒、反转录试剂盒、实时荧光定量试剂盒、Trizol试剂、二甲基亚砜(DMSO)购自Solarbio公司;噻唑蓝(MTT试剂盒、异硫氰酸荧光素标记的膜联素V/碘化丙啶(AV-FITC/PI)细胞凋亡检测试剂盒、双荧光素酶试剂盒均购自北京碧云天生物公司;LipofectamineTM2000转染试剂购自Invitrogen公司;载体质粒均由金瑞斯生物科技公司构建;酶标仪购自南京德铁实验设备公司。
1.2方法
1.2.1细胞培养 复苏H9C2细胞,使用含10% 胎牛血清(FBS)的DMEM培养液,在37℃、5%CO2、95%O2条件下培养;2 d换液1次,取处于对数生长期的细胞进行试验。
1.2.2细胞转染和分组 将心肌细胞随机分为两组,空白对照(Control)组:不做任何处理;单纯缺氧组:将心肌细胞用无血清低糖的DMEM培养基培养,置于持续充入95%N2+5%CO2的密闭培养箱中进行缺氧处理48 h。
将miR-con、miR-194、anti-miR-con、anti-miR-194、pcDNA和pcDNA-KLF15质粒转染到正常培养的心肌细胞中,记为miR-con组、miR-194组、anti-miR-con组、anti-miR-194组、pcDNA组和pcDNA-KLF15组;再将anti-miR-con组、anti-miR-194组、pcDNA组和pcDNA-KLF15组细胞进行缺氧处理,记为缺氧+anti-miR-con组、缺氧+anti-miR-194组、缺氧+pcDNA组和缺氧+pcDNA-KLF15组。
将miR-194质粒分别和pcDNA质粒、pcDNA-KLF15质粒共转染到正常培养的心肌细胞中再行缺氧处理,记为缺氧+miR-194+pcDNA组和缺氧+miR-194+pcDNA-KLF15组;将anti-miR-194质粒分别和si-con质粒、si-KLF15质粒共转染到正常培养的心肌细胞中再做缺氧处理,分别记为缺氧+anti-miR-194+si-con组和缺氧+anti-miR-194+si-KLF15组;转染均按照LipofectamineTM2000试剂盒进行操作。
1.2.3qRT-PCR检测细胞miR-194和KLF15 mRNA水平 用Trizol试剂提取总RNA,分别使用TaKaRa反转录试剂盒和荧光定量试剂盒,将RNA反转录为cDNA并避光配制反应体系,以β-actin为内参进行PCR扩增,循环条件为95℃ 30 s,60℃ 30 s;72℃ 30 s,共40个循环;60℃延长5 min。每个样品重复3次,采用2-△△Ct法分析GPX1 mRNA相对表达量。
1.2.4Western印迹检测蛋白表达水平 收集以上各组细胞,加入RIPA细胞裂解液于冰上裂解30 min;4℃,12 000 r/min离心15 min,取上清。BCA试剂盒测定蛋白样品浓度后进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE),电泳结束后,将蛋白电转至聚偏氟乙烯(PVDF)膜上,50 g/L脱脂奶粉室温封闭1 h;分别加入兔抗人KLF15多克隆抗体(1∶1 000)、兔抗人p-STAT3多克隆抗体(1∶1 000)、兔抗人STAT3多克隆抗体(1∶1 000)、兔抗人酶切caspase-3多克隆抗体(1∶1 000)、兔抗人CyclinD1多克隆抗体(1∶1 000)、兔抗人β-actin多克隆抗体(1∶1 000),4℃ 过夜;洗膜后,加入HRP标记的山羊抗兔IgG(1∶1 000),室温孵育2 h;TBST洗膜3次,10 min/次。后在暗室中曝光显影,再浸入定影,最后洗去残液晾干,将胶片用Quantity One凝胶分析软件处理,测定各组蛋白条带的吸光度,以目的条带和β-actin条带的比值作为蛋白表达水平,实验重复3次。
1.2.5MTT检测细胞增殖 在以上各组细胞培养至48 h时加入20 μl(5 g/L)的MTT溶液,继续孵育4 h;弃去多余培养基并加入150 μl DMSO振荡反应10 min,酶标仪检测490 nm处吸光度(A)值。细胞增殖存活率(%)=实验组A值/空白对照组A值×100%。
1.2.6流式细胞术检测细胞凋亡 取各组细胞,胰蛋白酶消化后4℃、300 r/min离心10 min,预冷磷酸盐缓冲液(PBS)洗涤后重悬。按照AV-FITC/PI细胞凋亡检测试剂盒说明书,分别加入5 μl AV-FITC和10 μl PI,轻轻混匀后室温避光孵育15 min,流式细胞仪检测激发波长488 nm和发射波长530 nm处的荧光强度,实验重复3次。
1.2.7荧光素酶报告基因检测实验检测miR-194对KLF15的靶向调控 TargetScan数据库显示KLF15 3'UTR区域有miR-324-5p结合位点。构建野生型和突变型基因靶点KLF15的3'UTR-荧光素酶表达载体(WT-KLF15和MUT-KLF15),取对数生长期心肌细胞H9C2接种于24孔板(1×103个/孔),待细胞生长至80 %汇合时,用LipofectamineTM2000分别将WT-KLF15和MUT-KLF15质粒转染到H9C2,同时分别转染miR-194质粒和miR-con质粒。依据说明书要求,使用荧光素酶报告基因检测仪进行双荧光素酶报告实验测定,实验结果以荧光素酶活性和Renilla活性的比值进行统计学分析。实验重复3次。
1.3统计学方法 采用SPSS22.0软件进行t检验、单因素方差分析。
2 结 果
2.1缺氧处理心肌细胞对miR-194和KLF15表达的影响 Western印迹检测结果显示,与Control组相比,缺氧组心肌细胞中KLF15蛋白表达水平显著降低(P<0.05)。可见,缺氧处理促进心肌细胞中miR-194的表达,抑制KLF15的表达。qRT-PCR检测结果显示,与Control组相比,缺氧组心肌细胞中miR-194的表达水平显著升高,KLF15 mRNA的表达水平显著降低(均P<0.05)。见图1,表1。

图1 检测心肌细胞中KLF15蛋白表达
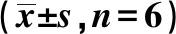
表1 缺氧处理心肌细胞影响miR-194和KLF15表达
2.2抑制miR-194表达促进缺氧处理心肌细胞存活和抑制细胞凋亡 Western印迹检测结果显示,与Control组相比,缺氧组心肌细胞中酶切caspase-3蛋白表达水平显著升高,CyclinD1蛋白表达水平显著降低(均P<0.05);与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞中酶切caspase-3蛋白表达水平显著降低,CyclinD1蛋白表达水平显著升高(均P<0.05)。qRT-PCR检测结果显示,与Control
组相比,缺氧组心肌细胞中miR-194表达水平显著升高,与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞中miR-194表达水平显著降低(均P<0.05)。MTT法检测结果显示,与Control组相比,缺氧组心肌细胞存活率显著降低(P<0.05),与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞存活率显著升高(P<0.05)。流式细胞仪检测结果显示,与Control组相比,缺氧组心肌细胞凋亡率显著升高(P<0.05);与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞凋亡率显著降低(P<0.05)。见图2、表2。可见,抑制miR-194表达促进缺氧处理心肌细胞存活和抑制细胞凋亡。


1~4:Control组,缺氧组,缺氧+anti-miR-con组,缺氧+anti-miR-194组图2 流式细胞仪检测心肌细胞凋亡
表2 转染anti-miR-194促进缺氧处理心肌细胞存活并抑制细胞凋亡
2.3miR-194靶向调控KLF15的表达 通过TargetScan数据库预测到KLF15与miR-194存在结合位点,见图3A。荧光素酶报告基因检测实验结果显示,相较于miR-con组,miR-194组WT-KLF15的心肌细胞H9C2荧光素酶活性显著降低(P<0.05);而MUT-KLF15的心肌细胞H9C2荧光素酶活性差异不显著(P>0.05)。见表3。Western印迹结果显示,相较于miR-con组(0.54±0.04),miR-194组(0.23±0.03)心肌细胞H9C2中KLF15表达显著降低(P<0.05);而相较于anti-miR-con组(0.49±0.05),anti-miR-194组(0.84±0.07)心肌细胞H9C2中KLF15表达显著升高(P<0.05)。见图3B可见,miR-194靶向调控KLF15的表达。
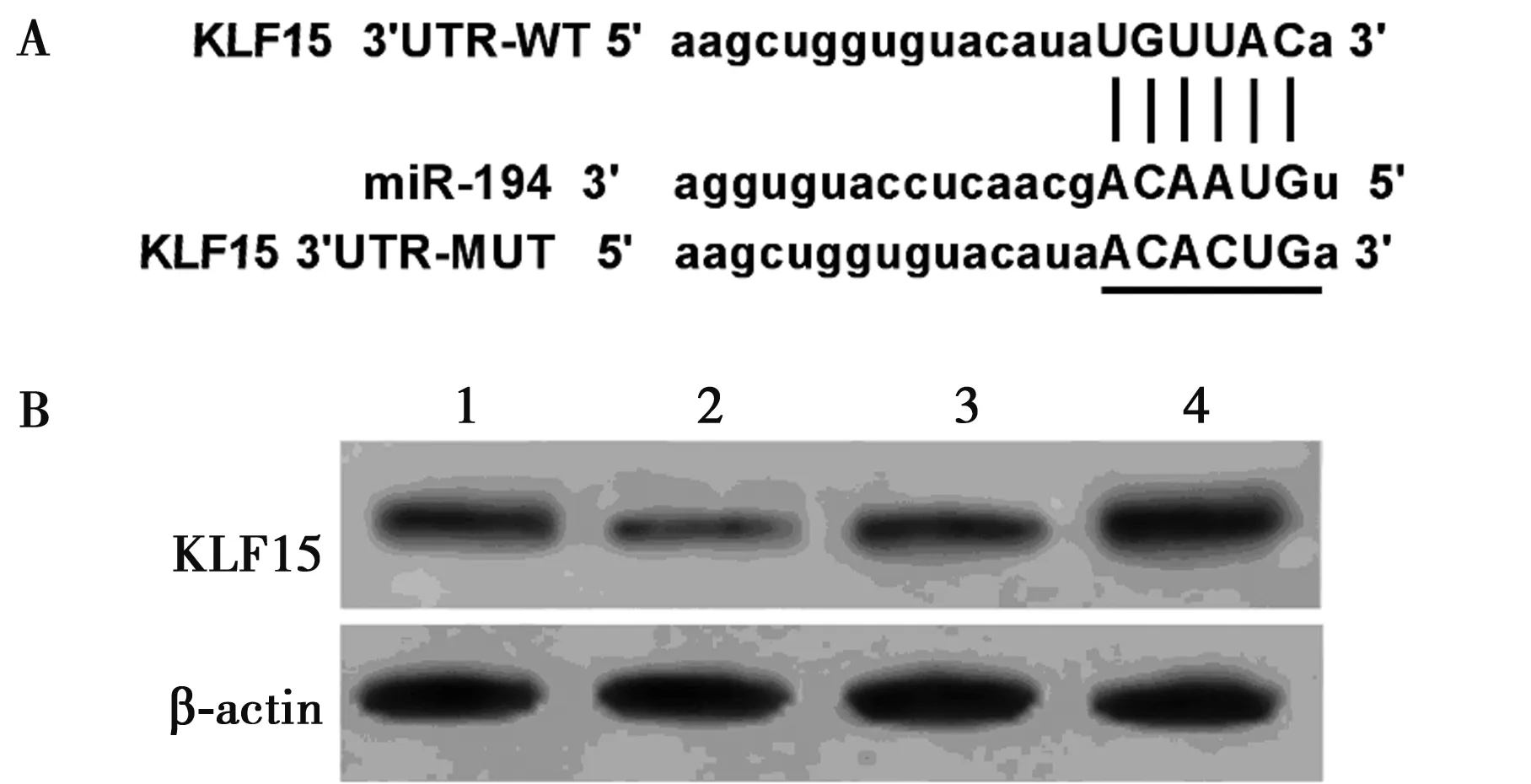
2.4过表达KLF15促进缺氧处理心肌细胞增殖和抑制细胞凋亡 Western印迹结果显示,与缺氧+pcDNA组相比,缺氧+pcDNA-KLF15组心肌细胞中KLF15和CyclinD1蛋白表达水平均显著升高(均P<0.05),酶切caspase-3蛋白表达水平显著降低(P<0.05)。MTT法检测结果显示,与缺氧+pcDNA组相比,缺氧+pcDNA-KLF15组心肌细胞存活率显著升高(P<0.05)。流式细胞仪检测结果显示,与缺
氧+pcDNA组相比,缺氧+pcDNA-KLF15组心肌细胞凋亡率显著降低(P<0.05)。可见,过表达KLF15促进缺氧处理心肌细胞增殖及抑制细胞凋亡。见表4,图4。
1~4:miR-con组,miR-194组,anti-miR-con组,anti-miR-194组图3 KLF15的3'UTR中含有与miR-194互补的核苷酸序列
表3 双荧光素酶报告实验
表4 过表达KLF15促进缺氧处理心肌细胞增殖和抑制细胞凋亡

1,2:缺氧+pcDNA组,缺氧+pcDNA-KLF15组图4 检测心肌细胞中KLF15蛋白表达
2.5沉默KLF15部分逆转抑制miR-194对缺氧处理心肌细胞的保护作用 Western印迹结果显示,与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞中KLF15和CyclinD1蛋白表达水平均显著升高(均P<0.05),酶切caspase-3蛋白表达水平显
著降低(均P<0.05);与缺氧+anti-miR-194+si-con组相比,缺氧+anti-miR-194+si-KLF15组心肌细胞中KLF15和CyclinD1蛋白表达水平显著降低,酶切caspase-3蛋白表达水平显著升高(均P<0.05)。MTT法检测结果显示,与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞存活率显著升高(P<0.05);与缺氧+anti-miR-194+si-con组相比,缺氧+anti-miR-194+si-KLF15组心肌细胞存活率显著降低(P<0.05)。流式细胞仪检测结果显示,与缺氧+anti-miR-con组相比,缺氧+anti-miR-194组心肌细胞凋亡率显著降低(P<0.05);与缺氧+anti-miR-194+si-con组相比,缺氧+anti-miR-194+si-KLF15组心肌细胞凋亡率显著升高(P<0.05)。见表5、图5。可见,沉默KLF15部分逆转抑制miR-194对缺氧处理心肌细胞的保护作用。
表5 沉默KLF15部分逆转降低miR-194对缺氧处理心肌细胞的保护作用
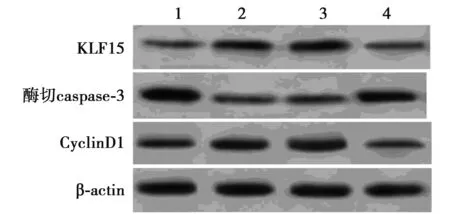
1~4:缺氧+anti-miR-con组,缺氧+anti-miR-194组, 缺氧+anti-miR-194+si-con组,缺氧+anti-miR-194+si-KLF15组图5 检测心肌细胞中KLF15蛋白表达
2.6miR-194调控KLF15对STAT3和p-STAT3的表达 Western印迹结果显示,与Control组相比,缺氧组心肌细胞中p-STAT3蛋白表达水平显著降低(P<0.05),而STAT3蛋白表达水平无显著差异(P>0.05);与缺氧+miR-con组相比,缺氧+miR-194组心肌细胞中p-STAT3蛋白表达水平显著升高(P<0.05),STAT3蛋白表达水平无显著差异(P>0.05);与缺氧+miR-194+pcDNA组相比,缺氧+miR-194+pcDNA-KLF15组心肌细胞中p-STAT3蛋白表达水平显著降低(P<0.05),STAT3蛋白表达水平无显著差异(P>0.05)。见图6、表6。可见,上调KLF15表达部分逆转miR-194对缺氧处理心肌细胞中p-STAT3蛋白表达的抑制作用。
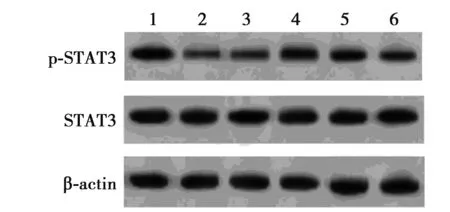
1~6:Control组,缺氧组,缺氧+anti-miR-con组,缺氧+anti-miR-194组,缺氧+anti-miR-194+si-con组,缺氧+anti-miR-194+si-KLF15组图6 检测心肌细胞中STAT3和p-STAT3的表达
表6 miR-194调控KLF15对STAT3和p-STAT3的表达
3 讨 论
急性心肌梗死是一种常见的心肌坏死型心血管急危重病,有发病急、病程短及死亡率高等特点,主要由冠状动脉急性或持续性缺氧所引发〔8〕。 miRNAs是心血管疾病新的标志物,急性心肌梗死、心肌病、心律失常和动脉血管硬化等疾病的病理改变早期有miRNA分子的参与,miRNA在其中发挥重要作用〔9〕。筛选缺氧性心肌病相关的miRNA,探讨miRNA的作用分子机制,对其诊断、治疗和预后具有指导意义。已有研究表明miR-182〔10〕、miR-19a-3p〔11〕等参与心肌细胞的缺氧坏死过程。miR-499可通过活化PI3K/Akt信号通路减少缺氧/复氧所诱导的心肌细胞凋亡〔12〕。miR-194通过调节IGF-1R/Akt信号通路抑制胶质瘤细胞增殖和糖代谢能力〔13〕;miR-194通过负向调控RBX1表达,从而抑制胃癌细胞的生长和转移,促进细胞凋亡和细胞周期的阻滞〔14〕。miR-194在黑色素瘤细胞中低表达,miR-194表达上调可促进黑色素瘤细胞凋亡并抑制黑色素瘤细胞的增殖,其机制可能与下调CyclinD1蛋白及上调酶切caspase-3蛋白表达有关〔15〕。缺氧会调控miR-194-1启动子的转录调控〔16〕。本研究结果表明,缺氧处理的心肌细胞中miR-194高表达,抑制miR-194表达促进缺氧处理的心肌细胞存活并抑制细胞凋亡。
Krüppel样因子KLF15是Cys2/His2锌指DNA结合蛋白的成员,在细胞的生长、分化、凋亡等生理过程发挥重要作用,研究发现KLF15抑制肺腺癌细胞的生长,可作为肺腺癌患者的治疗靶点和预后分子标志物〔17〕。KLF15通过上调CDKN1A/p21和CDKN1C/p57表达抑制胃癌细胞的细胞增殖〔18〕。KLF15过表达通过抑制结缔组织生长因子保护啮齿动物模型中β-氨基丙腈诱导的主动脉破裂〔19〕。KLF15通过调节细胞死亡和抑制Akt/ mTOR信号传导来预防异丙肾上腺素诱导的心脏肥大〔20〕。KLF15可通过反馈调节TGF-β,抑制CTGF及MRTF-A的作用,减轻心肌间质纤维化及心肌成纤维细胞表型转变,从而改善心功能〔21〕。然而KLF15在心肌细胞中的作用机制还尚未完全清楚,本研究结果显示,缺氧处理的心肌细胞中KLF15低表达,过表达KLF15促进缺氧处理的心肌细胞存活并抑制细胞凋亡。
周舟〔22〕研究发现caspase-3激活在缺氧诱导心肌细胞凋亡过程中具有重要作用,缺氧可上调心肌细胞caspase-3基因表达,而本研究结果显示,缺氧心肌细胞中酶切caspase-3蛋白的表达水平显著升高,而miR-194抑表达和KLF15过表达会使降低缺氧心肌细胞中酶切caspase-3蛋白的表达,证实了前人研究结果。而且有关miR-194与KLF15的调控关系及它们对缺氧诱导的心肌细胞凋亡的影响还鲜有报道,而本研究发现miR-194靶向负调控KLF15的表达。沉默KLF15部分逆转miR-194抑制表达对缺氧处理心肌细胞的保护作用;过表达KLF15部分逆转miR-194过表达对缺氧处理心肌细胞中p-STAT3蛋白表达的促进作用。
综上,抑制miR-194表达能降低缺氧诱导的心肌细胞损伤,其机制可能与调控KLF15及p-STAT3蛋白的表达相关,为心肌细胞损伤的治疗提供新思路和新靶点。
