系列C1化学分子与石墨炔表面相互作用的分子模拟
2021-05-20高红凤乔卫叶白红存
刘 喆, 高红凤, 王 强, 吴 勇, 乔卫叶, 白红存
(1.宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室 化学化工学院,宁夏 银川 750021;2.邢台学院 化学与化工学院,河北 邢台 054000)
C1化学在能源与环境领域中占有重要地位[1]。一般而言,C1化学是由1个碳原子组成的小分子物质为原料进行物质合成的化学,其稳态的化学物种主要包括CO2、CO、CH4以及由此衍生的CH3OH、HCHO和HCOOH等。以C1化学物种为原料,可以合成一系列基础化工产品。由此发展的C1催化、甲烷重整、生物质转化、合成气制取以及应用技术均与C1化学领域密切关联[2-5]。近几十年来化石加速消耗,导致CO2大量排放,被认为是温室效应显著增强的重要原因。因此,CO2减排和捕获利用成为能源环境方面的热点研究领域[6]。CO作为合成气和各类煤气的关键组分,在现代煤化工领域具有举足轻重的地位,被认为是合成一系列基本有机化工产品和中间体的重要原料[7]。氨、光气以及醇、酸、酐、酯、醛、醚、胺、烷烃和烯烃等绝大部分基础化工原料可通过CO为原料合成。CH4作为沼气、天然气、煤矿瓦斯的最重要组分,长期以来是重要的能源供给和化工原料。特别是,将CH4催化转化为合成气是利用CH4合成高附加值化工产品的重要途径[8]。此外,基于CO、CO2和CH4化学转化制备的CH3OH、HCHO和HCOOH等C1化学物质在能源、化工原料、食品、医药、纺织等国计民生领域均占据重要地位[9]。因此,C1分子的物理和化学行为以及反应特性,尤其是它们与催化和分离材料的表面作用一直是工业和工程化学的重要研究课题。
石墨炔是中国科学家李玉良等首次合成的新型碳同素异形体[10]。与石墨烯类似,石墨炔由碳元素组成,具备二维单原子厚度的规整有序孔道结构。又与石墨烯不同,石墨炔结构中除sp2杂化碳原子外,还包括sp杂化碳原子[11],因此,组成了化学键更为丰富的二维平面网络构型。石墨炔特殊的超大共轭结构、更多的C—C不饱和键以及良好的稳定性,使其在能源、催化、分离等诸多领域表现出显著的潜在应用优势[12-13]。例如,作为高效分离膜材料,石墨炔被认为在CO、CO2、CH4和CH3OH等C1化学体系分离中具备显著优势[14]。负载铁原子的石墨炔被认为是很有前途的CO2电还原制CH4和乙醇的电催化剂[15]。经金属Li掺杂的官能化石墨炔显示出良好的CO2捕获和封存潜力[16]。此外,负载不同金属(Sc、Ti等)后的石墨炔催化剂可实现CO高效氧化[17]。同时,石墨炔在无金属的低温条件下也可进行CO催化[18]。需要指出的是,在上述能源转化、催化、分离等过程中,C1小分子物质与石墨炔表面的吸附和界面相互作用是实现高效利用的关键步骤。由于没有获得这些分子与石墨炔的稳定吸附构象,因此二者之间的作用机制尚不明确。到目前为止,关于C1化学分子与石墨炔表面相互作用的系统研究未见报道。
笔者应用先进的密度泛函理论(Density functional theory,DFT)计算方法,系统研究6种C1分子(CH4、CO、CO2、CH3OH、HCHO和HCOOH)与石墨炔表面的相互作用。重点考察C1分子的不同吸附构象以及与石墨炔表面的不同作用位点,总结优势吸附的稳定性结构特征。通过基于C1分子与石墨炔之间非共价相互作用的定性和定量的描述,揭示分子间相互作用的内在机制。
1 模型与计算方法
选择如图1所示的正六角形石墨炔片段作为底物研究C1分子的吸附,其化学式表示为C66H18。该结构中心和6个角均为类似于苯的C6环,另有6个 C12环结构位于中心C6环周围。
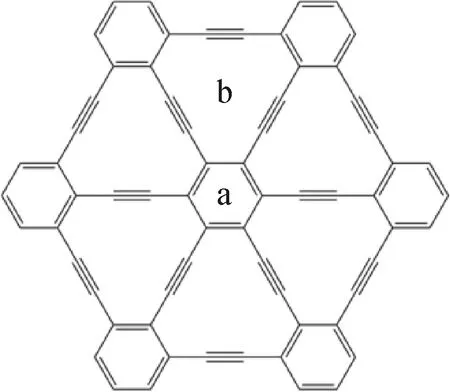
图1 石墨炔纳米片段的结构以及a和b吸附位点Fig.1 The structure of nano-scale graphyne and two adsorption sites a and b
研究中考虑了2种不同的吸附位点,分别为处于中心C6环的a吸附点和处于C12环中心的b吸附点。此外,对于每个吸附位点上的6种C1分子均考虑不同的吸附构象。以a吸附位点为例,6种C1分子共计考虑18种可能的吸附构象,并进一步通过计算确认不同吸附位点上各C1分子的最优吸附构象。这些在a和b位点吸附的不同构象经过几何构型优化后如图2所示。其中,3种CO吸附构象分别为:C—O键平行于底层(P型)、C—O键垂直于底层且碳原子靠近底层(S1型)和C—O键垂直于底层且O原子靠近底层(S2型)。2种CO2吸附构象分别为分子平行于底层(P型)和分子垂直于底层(S型)。2种CH4构象分别为:3个氢原子靠近底层(H3型)和1个氢原子靠近底层(H1型)。3种CH3OH吸附构象分别为:C—O键平行于底层(P型)、3个氢原子靠近底层(H3型)和C—O键垂直于底层(S型)。3种HCHO吸附构象分别为:分子平面平行于底层(P型)、分子平面垂直于底层且O原子靠近底层(S1型)和分子平面垂直于底层且2个H原子靠近于底层(S2型)。5种HCOOH吸附构象分别为:分子平面平行于底层(P型)、分子平面垂直于底层且2个氧原子靠近底层(S1型)、分子平面垂直于底层且2个氢原子靠近底层(S2型)、C—O单键垂直于底层且羟基靠近底层(S3型)和C—O单键垂直于底层且醛基靠近底层(S4型)。在b点吸附的吸附构象与a点类似。
所有结构优化和单点能采用基于密度泛函理论的量子化学自洽场分子轨道法计算得到[19]。使用的DFT交换相关势为BP86方法[20]。考虑到C1分子与石墨炔之间主要为非共价键结合,因此在BP86方法基础上增加了第三代色散校正(D3),以满足研究吸附体系中对弱相互作用的相对精确描述要求[21]。结构优化使用Gaussian09程序[22]中6-31G (d,p)高斯型基函数展开。为了进一步研究相互作用的物理本质,笔者还使用AMS (Amsterdam modeling suite)软件和TZP基组对吸附作用进行了能量分解分析(EDA)计算[23]。通过能量分解可将相互作用片段之间的形成能(Eform)分解为4个部分,即静电相互作用能(Eelstat)、轨道相互作用能(Eorb)、Pauli排斥能(Epauli)和色散校正能量(Edisp)。需要指出的是,能量分解分析是研究复合体系之间相互作用的重要方法,在多个吸附和主客体相互作用体系中得到成功应用[23-25]。
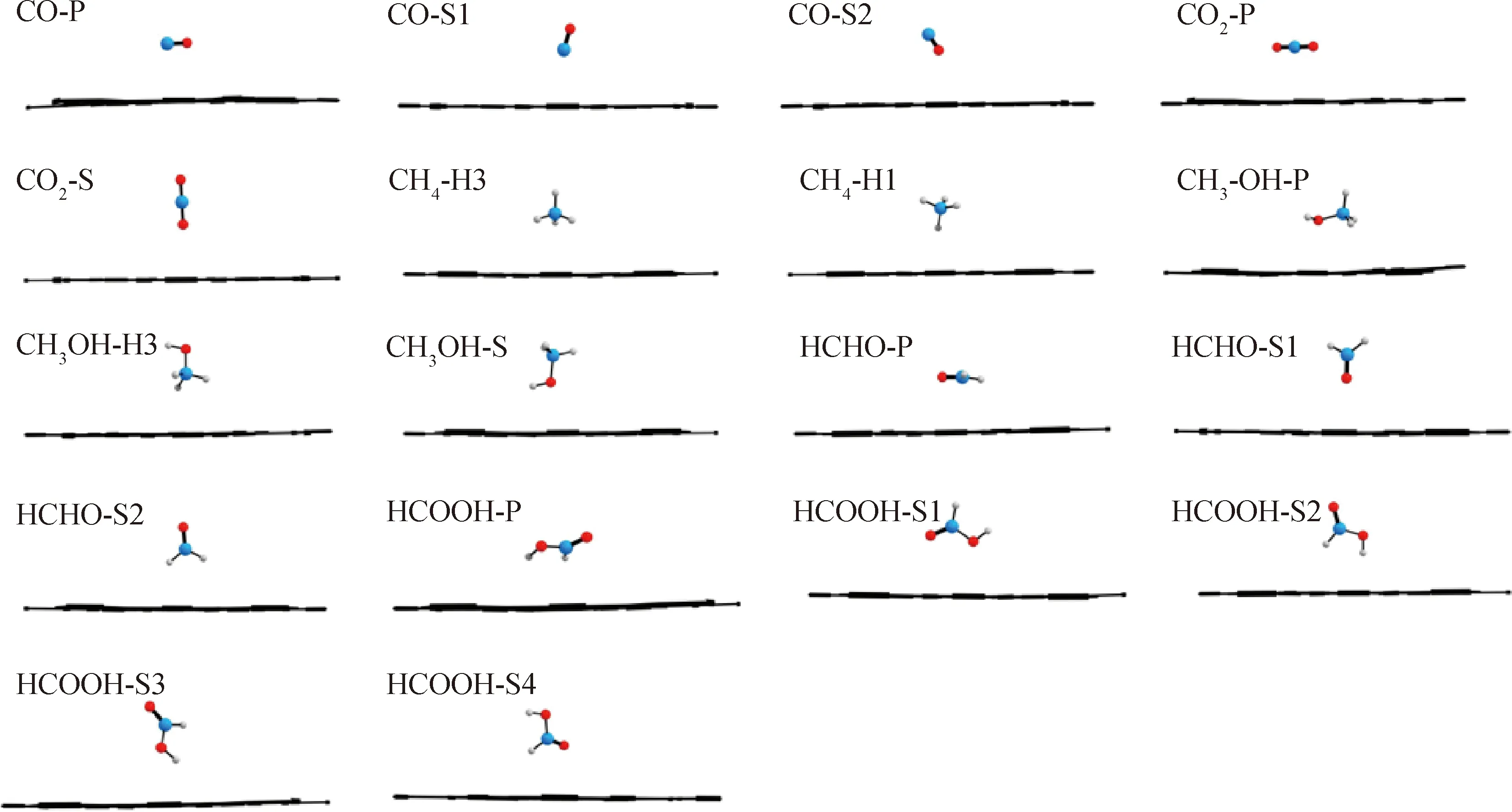
The blue, gray and red balls present carbon, hydrogen and oxygen atoms, respectively.P—Parallel conformation; S—Straight conformation; H—Hydrogen图2 6种C1分子在石墨炔a吸附点的不同吸附构象Fig.2 Different conformations of six C1 molecules at adsorption site a of the graphyne
2 结果与讨论
2.1 稳定吸附的结构特征
如图2所示,经完全几何构型优化得到的吸附构象与初始结构相比没有发生本质的变化。通过分析这些C1分子与石墨炔底层之间的距离,发现C1分子与石墨炔底层之间的原子相互作用最近距离大于0.22 nm,其中石墨炔的碳原子与C1分子中碳原子、氧原子和氢原子之间的相互作用距离分别为0.31~0.35、0.31~0.33、0.22~0.29 nm,考虑到碳、氧、氢元素的范德华半径分别为0.17、0.155、0.11 nm,因此,这些距离都主要集中在范德华距离作用范围内。表1为不同吸附位点和不同吸附构象的C1分子与石墨炔相互作用复合体系的相对能量和形成能。表1给出的形成能最大仅为43.51 kJ/mol,该数值明显小于共价键的键能范围[25]。因此,从相互作用距离和形成能两个角度分析,石墨炔和C1分子复合体系的两个片段间并无明显化学键合的特征[25],相互作用基本上处于范德华作用范围之内。这与之前报道的研究结果一致[26-28]。
由表1还可见,吸附位点和吸附构象的不同,导致各C1分子和石墨炔复合体系具有不同的相对能量。对于线性分子CO,其能量最低的优势吸附构象和能量最高的吸附构象均为C12环b吸附点表面,能量最低构象为以C—O键平行于底层(P型),而能量最高的吸附方式是以C—O键垂直于底层且氧原子靠近底层方式(S2型),两者能量差为6.73 kJ/mol。然而,同为线性分子的CO2的最低优势吸附构象和能量最高吸附构象分别处于b点和a点。前者为b点分子平行于底层(P型),后者为a点分子垂直于底层(S型),且前者吸附构象较后者能量低9.32 kJ/mol。可见,不同C1分子在石墨炔表面的优势吸附构象具有不同的结构特征。对于更复杂的非线型分子,CH4的能量最高和最低吸附方式均在a点,分别对应1个氢原子靠近底层(H1型)和3个氢原子靠近底层(H3型)。研究还发现,CO、CO2和CH4各不同吸附构象之间的能量差别不大(<9.32 kJ/mol)。这是由于这些C1分子具有相对较为简单的几何结构和势能面。
对于含氧的C1分子,CH3OH吸附的能量最低和最高吸附方式分别对应b点C—O键垂直于底层(S型)和a点(S型)。二者的能量差为9.7 kJ/mol,与CO2相应能量差非常接近。对于平面的HCHO分子,其能量最低和最高吸附方式分别对应b点分子平面平行于底层(P型)和a点分子平面垂直于底层且O原子靠近底层(S1型),二者能量差较大,达到28.76 kJ/mol。HCOOH是本研究中最复杂的C1分子,计算得到的最低优势吸附构象为b点分子平面垂直于底层且2个氢原子靠近底层(S2型);对于能量最高的吸附方式,则为a点分子平面垂直于底层且2个氧原子靠近底层(S1型)。这里优势构象较能量最高构象的能量差值达到25.67 kJ/mol。研究还发现,HCHO和HCOOH 2种C1分子,由于含有C、H、O 3种元素而具备更复杂的几何结构和势能面构造,因此HCHO和HCOOH分子在石墨炔表面不同吸附构象之间的能量差别较CO、CO2和CH4更为显著(>25 kJ/mol)。另外,除CH4外,所有C1分子最低优势吸附构象均出现在b点。
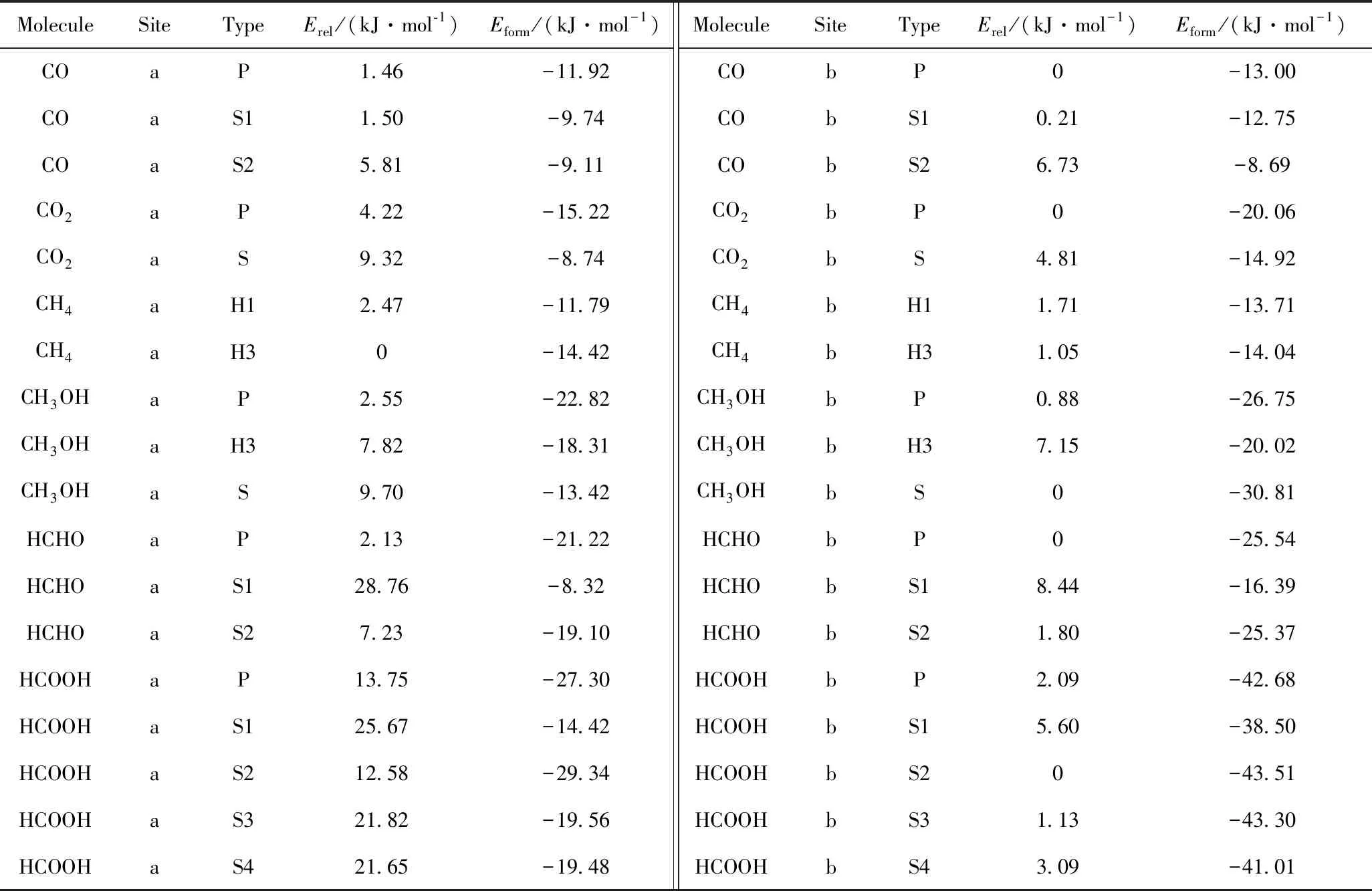
表1 不同吸附位点和吸附构象的C1分子和石墨炔复合体系相对能量(Erel)和形成能(Eform)Table 1 Relative energy (Erel) and formation energy (Eform) of C1 and graphene complex with different adsorption sites and conformations types
2.2 C1分子与石墨炔之间的吸附机理
为了定量描述吸附形成的复合物中石墨炔与系列C1分子之间的作用力强弱,对复合体系的形成能(Formation energy,Eform) 按公式(1)进行了计算,结果列于表1。
Eform=EA+B-EA-EB
(1)
式中:EA+B、EA和EB分别指吸附复合体系、吸附体系中C1分子和石墨炔底层分子的能量,kJ/mol。
由表1可见,所研究的全部36种吸附模型的形成能均为负值,即系列C1分子在石墨炔表面吸附形成复合体系的过程均为放热过程,在能量上对于吸附是有利的。这与CO2在金属掺杂的石墨炔表面吸附的放热过程有利于CO2捕集的结论相吻合[29]。不同的C1分子和不同的吸附构象对于形成能具有一定影响。CO分子表现出较小的形成能(8.69~13 kJ/mol),且不同吸附构象的形成能差距较小(<4.31 kJ/mol)。不同吸附构象的CO2和CH4的形成能分别为8.74~20.06 kJ/mol和11.79~14.42 kJ/mol,略大于CO形成能。对于CH3OH、HCHO和HCOOH,其最低优势构象的形成能明显增加,分别为30.81、25.54和43.51 kJ/mol。可见,这3种同时含有C、H和O 3种元素的C1分子与石墨炔表面结合得更为牢固。此外还发现,所有C1分子体系,能量最低的优势吸附构象和能量最高的吸附构象分别对应最大和最小的形成能,即最低优势吸附构象总是具有最大的形成能。
为了深入了解石墨炔与C1分子吸附相互作用的物理本质,笔者基于EDA研究了每类C1分子体系能量最低的优势构象和能量最高的构象[25]。通过能量分解将C1分子与石墨炔相互作用片段之间的形成能分解为静电相互作用能(Eelstat)、轨道相互作用能(Eorb)、Pauli排斥能(Epauli)和色散作用能(Edisp) 4个部分,结果见表2。
由表2可见,对于任意复合体系,计算获得的Epauli均为正值,即Pauli排斥作用对于复合体系片段之间表现为斥力作用,对体系的稳定化是不利的因素。这是由于Pauli作用源于电子在空间中占领轨道之间的相互作用,由于电子为费米子,故呈现排斥作用[30]。此外还发现,最低优势构象较能量最高构象具有更大的Epauli。对于Eelstat、Eorb和Edisp,任意复合体系获得的计算数据均为负值。因此,静电作用、轨道作用和色散作用在复合体系片段之间表现为吸引力,对复合体系的稳定化是有利的。笔者对吸附过程中起到积极作用的3种吸引力进行分析:对于CO吸附在石墨炔表面,最低优势吸附构象和最高吸附构象的Eelstat、Eorb和Edisp差值分别为7.12、6.98和3.93 kJ/mol,可见,静电作用和轨道作用的吸引力增强更加明显,是导致优势吸附构象能量更低的主要原因;对于CH3OH,静电作用和色散作用是导致构象之间能量差异的主要原因;对于HCOOH在石墨炔表面,最低优势吸附构象和最高吸附构象的能量差值分别为20.69、17.76和8.15 kJ/mol,所以能量最高和最低构象主要是由石墨炔和HCOOH之间的静电作用决定的;而对于CO2、CH4和HCHO吸附体系,色散作用的吸引力的增强更加明显,是导致优势吸附构象能量更低的主要原因。以上研究表明,对于不同C1分子多个构象间的能量差,静电作用、轨道作用和色散作用的决定因素是不同的。
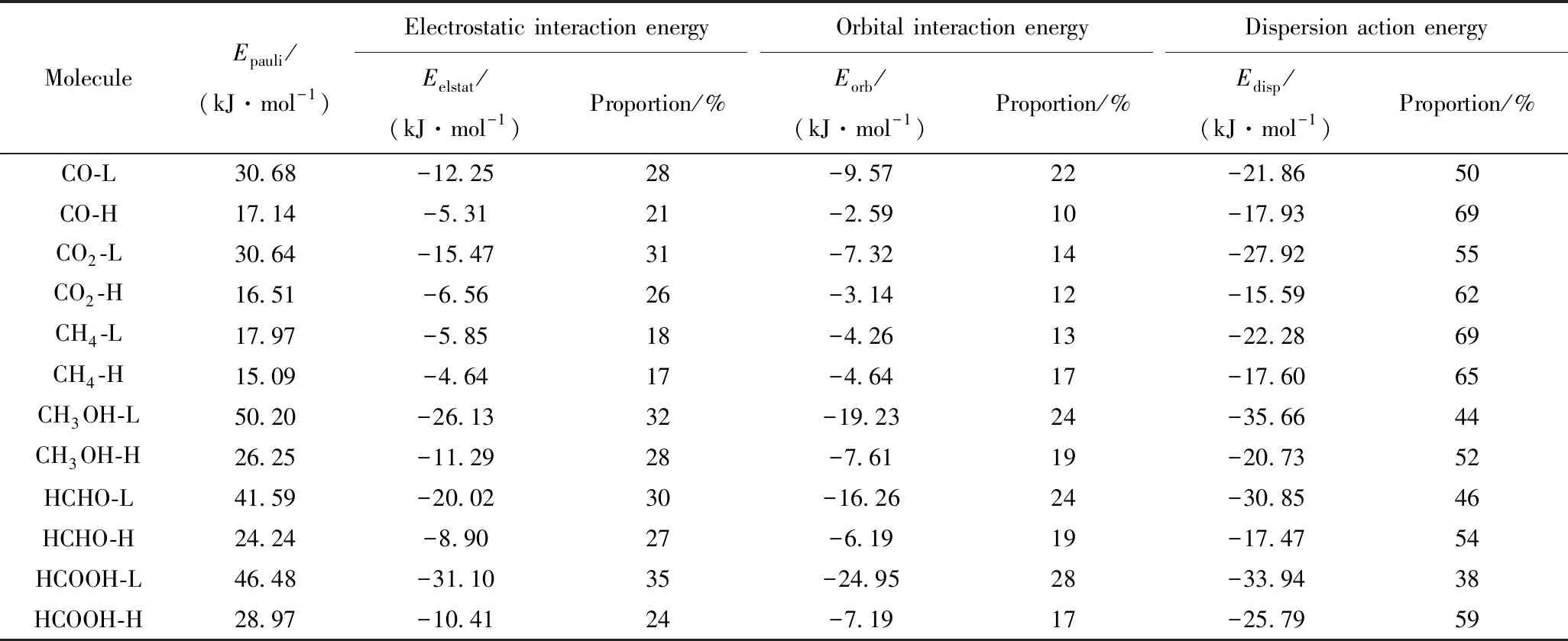
表2 C1分子吸附石墨炔能量最低和最高的复合体系能量分解分析的数值与所占比例Table 2 Energy decomposition analysis value and proportion of graphyne and C1 molecule composite system with the lowest and the highest energy
需要指出的是,尽管石墨炔与C1分子之间的静电作用、轨道作用和色散作用均表现为吸引力,但是它们各自对总体吸引作用的贡献是不同的。由表2可见,对于CH4、CO、CO2、CH3OH、HCHO和HCOOH 6种C1体系与石墨炔的相互作用,Edisp对于总体吸引作用(即三者相加总和)的贡献比例均为最高,其次为Eelstat,Eorb比例最低。因此,石墨炔与这些C1分子之间的相互作用主要由色散作用主导。尤其对于CO、CO2和CH43种体系,色散作用占总体吸引作用比例均超过50%。对于HCOOH最低优势吸附构象,静电作用的比例增加至35%,仅略低于色散作用38%的比例。这是由于HCOOH比其他C1分子表现出相对更强的分子极性,与石墨炔相互作用时更易实现片段间电荷迁移现象。
2.3 相互作用分析
由于吸附复合物中石墨炔与系列C1分子主要为非共价键相互作用,笔者基于约化密度梯度(Reduced density gradient, RDG)函数[31]对复合体系片段之间相互作用进行进一步研究。RDG是杨伟涛等提出研究弱相互作用的方法,已经成功应用于多种非共价相互作用的研究[25,32-34]。基于RDG分析,可凸显出相互作用的复合物中涉及到的片段之间相互作用的区域,可以直观地刻画复合物中相互作用的位点和类型。约化密度梯度函数可表示为:
(2)
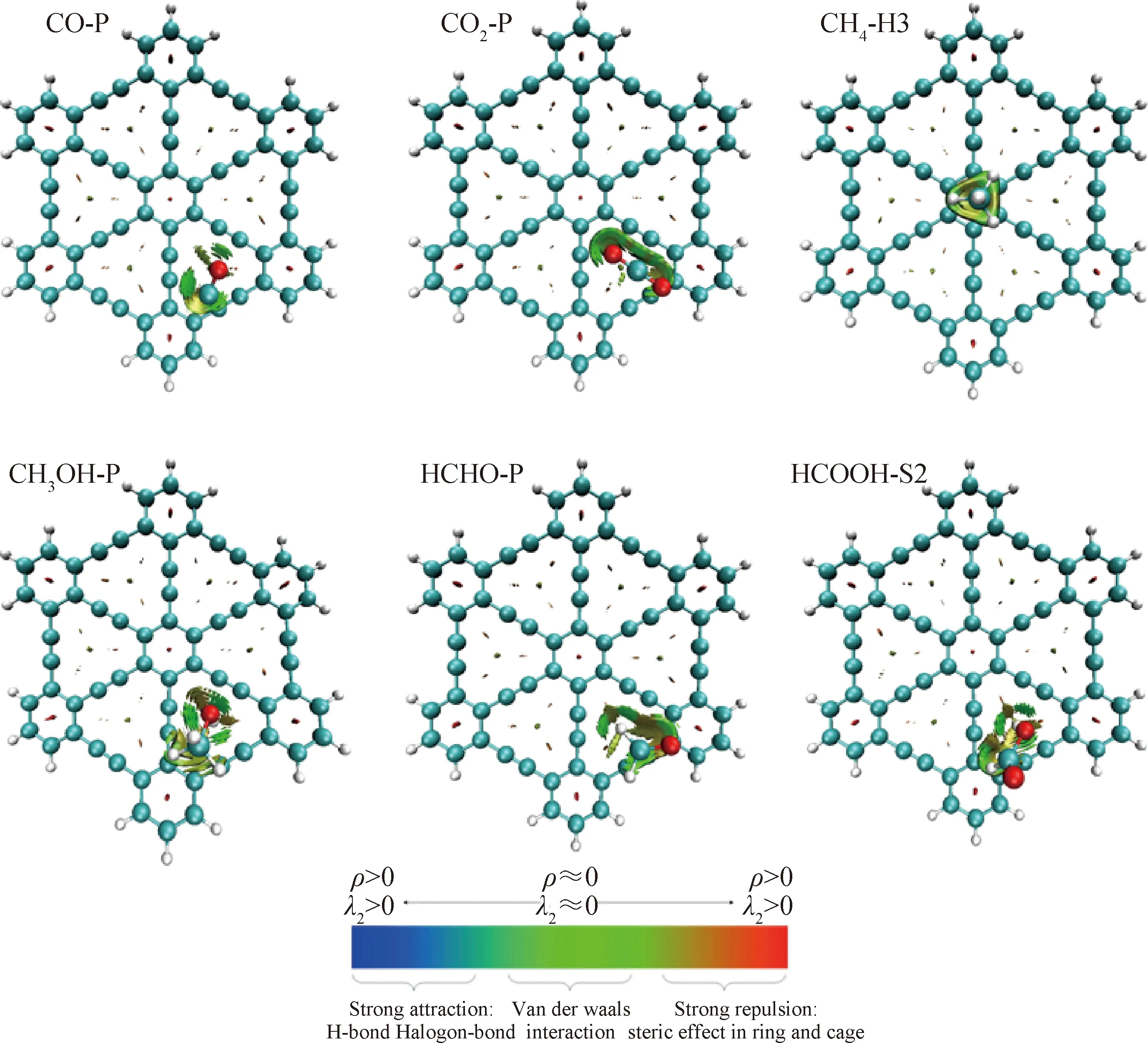
ρ—Interaction strength; λ2—Type of interaction; P—Parallel conformation; S—Straight conformation; H—Hydrogen图3 C1分子在石墨炔表面的RDG分析Fig.3 RDG analysis for C1 molecule on the surface of graphyne
由图3可见,石墨炔纳米片段中每个C6环的中心都存在红色作用区域,表明苯环中心的空间排斥作用,这与已报道的文献[35]相吻合。CH4的最低优势吸附构象出现在a点,CH4与石墨炔底层的作用位点主要集中在CH4向下的3个氢原子与石墨炔中心C6环之间的区域,且根据其颜色确定的表现类型为范德华作用力。这是由于单独的C—H、C—O、C=O 的键矩分别为1.3×10-30、2.5×10-30和7.7×10-30(C·m)。在笔者所研究的C1分子中,除了CH4分子外,其他C1分子中都含有C—O或者C=O。键矩较大时更容易在外界分子电场作用下进一步极化,形成较强的局部正负电荷中心。C12环中的炔键碳本身会形成局部的正负电荷中心,所以更容易与键矩较大的C—O或者C=O产生静电相互作用。而石墨炔基中的C6环中正负电荷中心带电量可能比较少,所以更倾向于以范德华力与C1分子作用。从表2可以看出,CH4-L与CH4-H体系中的色散作用能占比最高,而静电相互作用能比例最小。这说明键矩较小的CH4分子更倾向于选择范德华力作用原子数较多的C6环位置进行吸附,有利于体系的稳定。相比之下,其他C1体系(CO、CO2、CH3OH、HCHO和HCOOH)与石墨炔相互作用优势构象在b点,所以静电相互作用能的比例都比CH4分子的要高。因此,这些分子与石墨炔纳米片之间主要为C1分子与C12环的作用。根据图3显示的颜色,这些分子与石墨炔纳米片之间也主要为范德华作用。这与前文根据能量分解获得的结论是一致的。值得注意的是,这些C1分子在b点的作用区域较CH4与a点C6环的范围更大。尤其CH3OH几乎与C12环中每个碳原子均存在相互作用区域,这可能是导致CH3OH最低优势吸附构象具有最大Edisp而CH4体系Edisp较小的主要原因。
3 结 论
应用密度泛函理论计算方法,系统研究6种C1分子(CH4、CO、CO2、CH3OH、HCHO和HCOOH)与石墨炔表面的相互作用。主要结论如下:
(1)考虑不同吸附构象的C1分子在石墨炔纳米片a和b 2个位点的吸附,通过复合体系几何结构优化进而获得的相对能量,确定了各C1分子在石墨炔表面优势吸附构象的结构特征。研究发现,除CH4外,其他C1分子的优势吸附构象均出现在石墨炔大孔C12环处。
(2)石墨炔和C1分子复合体系形成能的计算结果表明,系列C1分子在石墨炔表面吸附形成复合体系的过程在能量上均是有利的。基于能量分解分析发现,石墨炔与优势构象的C1分子之间的相互作用主要由色散作用主导,其次为静电作用和轨道相互作用。
(3)约化密度梯度函数(RDG)分析直观地刻画了C1分子与石墨炔纳米片之间相互作用的区域和类型。相互作用颜色显示,C1分子与石墨炔纳米片之间主要为范德华作用,与能量分解的结论一致。
