Open late: neuronal membrane disruption late in traumatic brain injury
2021-04-29MartinaHernandezMiJinChoAudreyLafrenaye
Martina L. Hernandez, MiJin Cho, Audrey D. Lafrenaye
Membrane disruption is a pathology in which the cellular phospholipid bilayer, which is the cell’s primary defense from the extracellular environment, is compromised. Some neurons that are impacted by membrane disruption experience cell death, and others demonstrate delayed disruption, providing an opportunity for therapeutic intervention. This pathology is visualized using infusions of cell impermeable tracers, such as dextran, calcein and propidium iodide followed by microscopic analysis (Geddes et al., 2003a, b; Singleton and Povlishock, 2004;Farkas et al., 2006; Whalen et al., 2008; Cullen et al., 2011; Lafrenaye et al., 2014; Levine et al., 2016; Hernandez et al., 2019; LaPlaca et al.,2019; Prado and LaPlaca, 2020), and therefore little is known about membrane disruption clinically. However, membrane disruption has been well documented to occur in various preclinical models of traumatic brain injury (TBI).Membrane disruption was initially referred to as “mechanoporation,” which is a specific subset of cellular membrane disruption that occurs at the time of mechanical insult. In the mid-1990s, the LaPlaca group imaged neuronal mechanoporationin vitroimmediately following fluid shear stress injury of NT2-N human cells and found significant levels of lactate dehydrogenase, a ubiquitously expressed intracellular enzyme, in the extracellular fluid minutes following injury (LaPlaca et al.,1997). Further research found that cultured rat neurons subjected to biaxial stretch injury showed increased levels of cell impermeable tracer uptake based on the size of the tracer and the degree of strain used, suggesting that membrane disruption is non-uniform and directly linked to the severity of injury (Geddes et al., 2003a). Disruption of the plasmalemma was also shown to result in increases of intracellular calcium within minutes, which can trigger the release of proteases that cleave proteins needed to transport vesicles and endosomes to the membrane surface for repair and cellular packaging (LaPlaca et al., 1997;Geddes et al., 2003b). As a result, neuronal mechanoporation had been speculated to lead to dysregulated ionic flux, apoptosis, and radical oxidative damage often assessed in TBI and spinal cord injuries. In fact, some studies found correlations between mechanoporation and cell death following focal brain injury (Whalen et al., 2008; LaPlaca et al., 2019).
The Povlishock group also identified mechanoporated neuronal axons and soma following a diffuse TBI using the central fluid percussion injury (CFPI) model. Upon induction of CFPI in Sprague-Dawley rats, signs of mechanoporated neurons were observed as early as five minutes and as late as eight hours post-injury in various brain regions (Singleton and Povlishock, 2004). However, in this diffuse model of TBI, most of the mechanoporated and acutely membrane disrupted neurons did not demonstrate the traditional signs of death;rather, they appeared ultrastructurally normal(Singleton and Povlishock, 2004; Lafrenaye et al., 2012). We recently recapitulated the finding of little to no cell loss in the region demonstrating membrane disruption following CFPI, indicating that diffuse membrane disruption may not progress to cell death as was found in focal lesions (Hernandez et al.,2019).
Importantly, a subset of membrane disrupted neurons were found to reseal their plasmalemma following injury by multiple groups. Using shear injuryin vitro, the LaPlaca group found that some mechanoporated neurons were no longer permeable minutes post-injury (Geddes et al., 2003a; Cullen et al.,2011).In vivostudies using Sprague Dawley rats in which 10 kDa dextran conjugated to green and red fluorophores were infused into the lateral ventricle pre- and post-injury also found a subset of neurons containing only the preinjury administered dextran, indicating that they had resealed their membranes (Farkas et al.,2006; Lafrenaye et al., 2014). Further studies also found that not only were mechanoporated neurons capable of membrane resealing, but there was also a delayed membrane disrupted population of neurons that only contained the post-injury administered dextran and some neurons that displayed enduring membrane disruption, in which they contained both pre- and post-injury administered dextran(Lafrenaye et al., 2014). Additionally, these enduring membrane disrupted neurons were found to be correlated to secondary insults,such as elevations in intracranial pressure and appeared to be the subpopulation of membrane disrupted neurons that progressed to cell death following diffuse TBI exacerbated by secondary intracranial pressure elevations(Lafrenaye et al., 2014). Such findings demonstrate that, while some mechanoporated neurons continued to demonstrate membrane disruption, other neuronal membranes could reseal following injury. Moreover, acute resealing of mechanoporated rat neurons is Ca2+regulated in which resealing was found to be significantly reduced either without extracellular calcium or when intracellular calcium was chelated (Prado and LaPlaca,2020). This link between membrane resealing,or lack thereof, and recovery or cell death following injury could provide opportunities for therapies that leverage these resealing potentials in those injured neurons.
These studies, however, investigated mechanoporation and acute membrane disruption occurring within hours of injury, timing which limits the therapeutic potential of targeting neuronal membrane disruption. Additionally, the possibility of active membrane disruption occurring days to weeks following injury remained unknown.Therefore, Hernandez et al. (2019) evaluated an extended temporal profile of membrane disruption following diffuse TBI using the CFPI model in Sprague-Dawley rats. To visualize actively membrane disrupted cells in layers V and VI of the lateral neocortex, a membraneimpermeable fluorescently-tagged dextran was administered intracerebroventricularly 2 hours prior to sacrifice at 6 hours, 1 day, 3 days, 2 weeks, and 4 weeks post-injury. This strategy allowed the cell-impermeable dextran to percolate through the parenchyma of the neocortical region of interest, indicating those cells that had disrupted membranes at the time of sacrifice. Diffuse neuronal membrane disruption in the lateral neocortex was found to occur in two temporal waves following CFPI,a subacute wave occurring from 6 hours to 3 days post-injury, and a later wave occurring 2 and 4 weeks following injury (Hernandez et al.,2019). This study was the first to demonstrate active neuronal membrane disruption weeks following injury. Identification of a later secondary wave of membrane disruption drastically increases the potential therapeutic window for intervention and treatment of brain injury-induced membrane disruption from hours to weeks and possibly beyond (Figure 1). Additionally, the percent of membrane disrupted neurons at 1 week post-CFPI was indistinguishable from sham levels, indicating a potential for membrane resealing at a much later timepoint than previously thought.
In addition to finding multiple temporal waves of membrane disruption, Hernandez et al. also observed a subset of membrane disrupted neurons that lacked expression of the panneuronal marker, neuronal nuclei (NeuN)(Hernandez et al., 2019). NeuN was identified in 1992 and is commonly used as a panneuronal marker. It functions in alternative splicing of various proteins including Numb,which is important for neuronal development and differentiation, leading to the notion that loss of NeuN following injury could be linked to an ameliorative developmental reversion(Duan et al., 2016). Glial cells also do not express NeuN; previous studies have shown that glial cells, such as astrocytes, could be susceptible to membrane disruption (Cullen et al., 2011; Levine et al., 2016). Therefore,traditional markers of the primary glial cell types, glial fibrillary acidic protein for astrocytes, adenomatous polyposis coli (APC/CC-1) for oligodendrocytes, ionized calciumbinding adaptor molecule 1 for microglia, and neuron-glia antigen 2 (NG2) for NG2 cells,were immunofluorescently assessed for signs of membrane disruption at all time points following CFPI. However, none of the glial cell types demonstrated signs of membrane disruption (Hernandez et al., 2019). Similarly,another study investigating acute membrane disruption following focal brain injury also found no signs of astrocyte membrane disruption(LaPlaca et al., 2019). While these findings differ from the results in thein vitrostudies,in which astrocytic membrane disruption was observed, these discrepancies may be a result of injury modality and substrate (Cullen et al.,2011; Levine et al., 2016). Ultimately, however,it was found that the NeuN–membrane disrupted population was unlikely to be glial in nature rather, were likely neurons lacking NeuN expression.
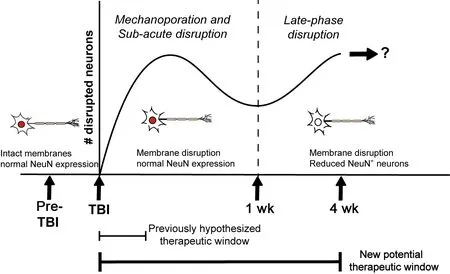
Figure 1|Timeline of membrane-disrupted neurons and coordinating therapeutic windows.
Upon further investigation, a NeuN–membrane disrupted population of neurons was observed to be present at all post-injury time points and were found to be significantly increased at 2 weeks post-CFPI (Hernandez et al.,2019). However, protein assessments did not reveal differences in NeuN expression in the lateral neocortex at any time following injury. This result indicates that the changes in NeuN could be occurring within a small subpopulation that would not be discernable with an evaluation of overall NeuN expression within the cortex. To interrogate this possibility,immunohistochemistry paired with hematoxylin and eosin counterstaining was used to quantify the percentage of NeuN expressing neurons within layers V and VI of the lateral neocortex.Interestingly, a NeuN–subpopulation of neurons was consistently observed in both sham and injured animals regardless of time points. This suggests there is a consistent subpopulation of NeuN–neurons within the lateral neocortex, a finding not previously reported. There are neuronal subtypes, such as subsets of interneurons, that do not express NeuN, which could represent this consistent NeuN–subpopulation (Duan et al., 2016).Additionally, the NeuN A60 antibody was used in this study and requires phosphorylation of at least one site on the N-terminus of NeuN to properly label (Duan et al., 2016). Alterations in the phosphorylation state of NeuN in various neuronal subpopulations within the cortex may also explain the consistently present NeuN–subpopulation found by Hernandez et al. (2019).In these cases, these NeuN- neurons may be representative of a neuronal subpopulation that is more vulnerable to late-phase membrane disruption following TBI. Alternatively, this NeuN–subpopulation could be indicative of a regenerative subset of neurons. A previous study found that a medullary respiratory neuronal subpopulation lost NeuN expression following axonal transection and that this loss of NeuN was associated with subsequent axonal regeneration (Darlot et al., 2017). This finding suggests that the NeuN–population could be reverting to a growth-permissive immature state representing a potential compensatory mechanism that may be associated with enhanced membrane resealing following diffuse TBI (Figure 1). These are possibilities that require future investigation and are currently being explored in follow-up studies.
The findings from Hernandez et al. (2019)highlight the heterogeneity of TBI-induced neuronal membrane disruption beyond mechanoporation and extend the potential therapeutic window for this pathology further than previously expected. Taken together, membrane disruption exhibits a biphasic nature through a broad post-injurytimeframe and involves phenotypically different subpopulations, specifically in relation to NeuN expression, following injury. Importantly,in both acute and late-phase membrane disrupted populations neurons maintain the ameliorating capacity for membrane repair which could serve as the framework for therapeutic intervention. The study indicates that a greater understanding of disruptions to neuronal integrity could lead to the discovery of treatments that reduce morbidity associate with diffuse pathology.This work was supported by the National Institutes of Health-National Institute of
Neurological Disorders and Stroke grant (No.1R01NS096143).
Martina L. Hernandez, MiJin Cho,Audrey D. Lafrenaye*
Department of Anatomy and Neurobiology, Virginia Commonwealth University, Richmond, VA, USA
*Correspondence to:Audrey D. Lafrenaye, PhD,audrey.lafrenaye@vcuhealth.org.https://orcid.org/0000-0001-5986-2228(Audrey D. Lafrenaye)
Date of submission:September 30, 2020
Date of decision:December 4, 2020
Date of acceptance:March 3, 2021
Date of web publication:April 23, 2021
https://doi.org/10.4103/1673-5374.313029
How to cite this article:Hernandez ML, Cho M,Lafrenaye AD (2021) Open late: neuronal membrane disruption late in traumatic brain injury.Neural Regen Res 16(12):2409-2410.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Inhibition of extracellular vesicle pathway using neutral sphingomyelinase inhibitors as a neuroprotective treatment for brain injury
- Neuroglobin and neuroprotection: the role of natural and synthetic compounds in neuroglobin pharmacological induction
- Potential effects of mesenchymal stem cell derived extracellular vesicles and exosomal miRNAs in neurological disorders
- Epidural electrical stimulation for spinal cord injury
- Localization of the hydrogen sulfide and oxytocin systems at the depth of the sulci in a porcine model of acute subdural hematoma
- Lithium beyond psychiatric indications: the reincarnation of a new old drug