促炎性细胞因子调控自噬与神经系统变性疾病研究进展
2021-04-26舒俊张丽
舒俊,张丽
复旦大学附属华东医院神经内科,上海200040
自噬是真核细胞内受损的蛋白质或细胞器通过溶酶体途径及时被降解,降解的产物再重新利用的一种生理过程,自噬在维持细胞稳态、细胞生长和自我更新方面发挥重要作用[1]。但过度自噬可能导致细胞死亡,即自噬性细胞死亡或II 型程序性细胞死亡[2]。既往研究显示自噬功能异常与神经系统变性疾病、肿瘤、免疫和炎症性疾病等多种疾病密切相关[3]。了解自噬的调节机制有助于自噬的精准调控,为疾病提供新的治疗方向。
机体受到损伤或病原体侵入时,先天和适应性免疫细胞产生促炎性细胞因子,帮助机体及时修复损伤,对全身免疫和炎症反应有重要影响[4]。促炎性细胞因子在阿尔茨海默病(Alzheimer's disease,AD)和帕金森病(Parkinson's disease,PD)等神经系统变性疾病发病机制中发挥重要作用。近年来的研究发现促炎性细胞因子参与自噬的调节过程,并借助调节自噬在肿瘤、炎症与免疫等多种疾病进展过程中发挥重要作用。而在神经系统变性疾病中促炎性细胞因子对自噬调节作用的相关研究较少。研究促炎性细胞因子与自噬之间的相互作用有助于更好地理解疾病尤其是神经系统变性疾病的发生和进展机制,从而制定更有效的治疗策略。本文主要综述了自噬过程及其调节机制;自噬和神经炎症反应与神经系统变性疾病;白介素-1(interleukin-1,IL-1)家族、IL-6、肿瘤坏死因子(tumor necrosis factor,TNF)等促炎性细胞因子对自噬调节的相关研究进展,为深入研究提供参考。
1 自噬过程
在哺乳动物细胞中,自噬主要可以分为微自噬、巨自噬和分子伴侣介导的自噬3 种类型[5]。本文所说的自噬指的是巨自噬,即受损的细胞器或蛋白质由双层膜的囊泡包裹,运送至溶酶体被降解的过程。自噬可简单划分为3 个过程,即自噬小体的形成、自噬溶酶体形成和自噬小体降解。其中自噬小体形成是整个自噬过程的关键步骤,涉及多个由自噬相关蛋白组成的复合物。微管相关蛋白轻链3-II(microtubule-associated protein light chain,LC3-II)或LC3-II/LC3-I 常作为自噬形成的标识物。见图1。
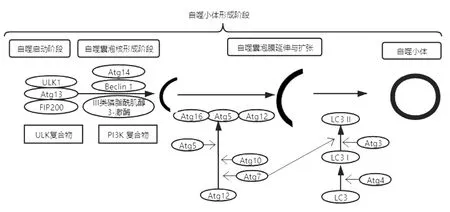
图1 自噬小体形成的过程[6-8]
2 自噬调节
研究显示多种蛋白或信号通路参与了自噬的调控,且这些信号通路可进行相互交叉调节。哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是自噬过程中的重要调节蛋白。在营养充足的条件下mTOR 与ULK1/2复合物结合,抑制自噬启动;反之则诱导自噬启动[9]。mTOR可受到多种信号通路调控,目前研究最多的是以下3 条通路。见图2。

图2 自噬相关通路
2.1 AMPK-mTOR1 信号通路 细胞能量不足时激活腺苷一磷酸活化蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK),活化的AMPK 磷酸化结节性硬化复合物1/2(tuberous sclerosis complex 1/2,TSC1/2)导致mTOR1失活,从而诱导细胞自噬水平的提高[10]。AMPK 也可直接激活ULK1 复合物,上调细胞自噬水平[11]。
2.2 Class I PI3K/Akt/mTOR1 信号通路 Class I PI3K 能在细胞中产生磷脂酰肌醇-3,4,5-三磷酸[phosphatidylinositol(3,4,5) -triphosphate,PIP3],PIP3 激活丝氨酸/苏氨酸激酶(serine/threonine kinase,Akt),进而抑制TSC1/2 和细胞自噬[12]。另外,磷酸酶和张力蛋白同源物(phosphatase and tensin homology deleted on chromosome ten,PTEN)可以抑制Class I PI3K 的活性,诱导细胞自噬水平上调[13]。
2.3 RAS/RAF/MEK/ERK 信号通路RAS/RAF/MEK/ERK 信号通路可以调节细胞凋亡、自噬和衰老等多种细胞功能。转化生长因子- 1 的岩藻糖基化修饰可通过PI3K/Akt 和RAS/RAF/MEK/ERK 信号通路增强卵巢癌细胞的自噬通量,为卵巢癌的靶向治疗提供了新的方向[14]。
3 自噬与神经系统变性疾病
自噬功能紊乱是AD 和PD 等神经系统变性疾病发生和进展的关键分子机制之一。自噬小体形成受阻[15]、自噬小体与溶酶体融合障碍或溶酶体降解途径受损[16-17]等,会导致细胞不能及时清除错误折叠或淀粉样蛋白(amyloid-protein,)淀粉样斑块、蛋白和突触核蛋白等病理性聚集,产生炎症反应和神经毒性,从而引起AD 和PD 等神经系统变性疾病的发生和进展。研究显示AD 早期患者脑内的自噬启动相关蛋白Beclin-1 表达量下降,且转基因AD 小鼠模型提示Beclin-1 表达减少,导致聚集和神经元损伤,而使用慢病毒载体过表达Beclin-1 可明显减少细胞内外聚集[18]。Beclin-1 的乙酰化修饰可导致自噬通量受损,参与AD 进展[19]。PD 相关的基因突变可导致溶酶体功能障碍,影响 突触核蛋白降解,从而促进其蓄积和聚集[20]。虽然许多研究显示提高自噬水平可减少细胞毒性蛋白的蓄积,延缓疾病进展[21],但过度或不恰当的激活自噬也可能造成神经元损伤[2],因此如何使自噬控制在合适的水平仍是未来研究的难点。
4 神经炎症与神经系统变性疾病
神经炎症反应是一种宿主防御系统,可在感染或脑损伤等应激条件下激活[22-24]。适当的炎症反应有助于机体及时修复损伤[25],但慢性持续的神经炎症反应会引起小胶质细胞过度活化和促炎性细胞因子持续释放,最终导致突触变性和神经元死亡[26]。研究证实神经炎症反应在AD 和PD等多种神经系统疾病的发病机制中起着关键作用[27-29]。在PD 患者中,外周血中的促炎性细胞因子IL-6 和IL-17A 水平升高,且与疾病严重程度相关[30],脑脊液中TNF-升高与疾病快速进展相关[31]。促炎性细胞因子IL-1、IL-6 和TNF-水平升高与和蛋白聚集相关,参与AD 疾病进展过程[32-33]。近年来的研究发现自噬在神经炎症调控机制中发挥重要作用[34]。自噬调节神经炎症主要通过降解诱导炎症反应产生的底物(如病原体或受损细胞器)和抑制促炎性细胞因子的产生及分泌,从而减轻神经炎症反应[35]。未来需要进一步研究神经系统变性疾病中自噬与神经炎症间的调控机制,从而制订更为有效的抑制神经炎症方案。
5 促炎性细胞因子调控自噬研究
自噬和炎症是两个重要的生物学过程,与机体生理和病理状态相关。各种细胞因子在自噬调节过程中也发挥重要作用,自噬与细胞因子之间的相互作用可能是协调先天免疫系统和适应性免疫系统活性的基本机制。
5.1 IL-1 家族 IL-1 家族的促炎性细胞因子主要包括IL-1、IL-1、IL-18、IL-33 和IL-36,在机体炎症和免疫过程中发挥重要作用[4]。这些细胞因子一般在炎症的早期释放,并且诱导机体进一步产生炎症反应,因此也被称为“警报蛋白”[36]。
IL-33 和IL-36 是新发现的IL-1 家族成员,可以在多种细胞组织类型中表达。IL-33 主要在星形胶质细胞和小胶质细胞中表达,但它在神经系统中的作用尚存争议。一些研究显示在脑出血[40]、脑外伤[41]和癫痫[42]等小鼠模型中,IL-33 通过抑制细胞自噬和炎症反应发挥神经保护作用。但最新的研究发现IL-33 可诱导海马区小胶质细胞活化,促进促炎性细胞因子释放,参与认知损害进展[43]。IL-36 通过调节自噬,从而在抵抗病毒和细菌等微生物感染过程中发挥免疫保护作用。IL-36 有IL-36、IL-36和IL-363 种存在类型。在甲型流感病毒感染早期,IL-36治疗可能通过Akt/mTOR 通路抑制自噬、促进细胞凋亡,控制病程进展[44]。在结核感染中,通过促进溶酶体形成,抑制细胞内结核分枝杆菌的生长,增强巨噬细胞抑菌能力[45]。IL-33 和IL-36 都是新发现的促炎性细胞因子,目前关于它们对自噬调节的研究较少,未来需要进一步的研究。
5.2 IL-6 IL-6 是一种炎症自分泌/旁分泌的多功能细胞因子,在许多生理和病理过程中起着重要作用。在胶质母细胞瘤中,IL-6 通过p-STAT3-MIR155-3p-CREBRF-CREB3-ATG5途径诱导细胞自噬,促进肿瘤生长;而IL-6 受体阻断剂可减少自噬,促进细胞凋亡,为肿瘤治疗提供潜在靶点[46]。在大动脉炎中,IL-6 通过激活JAK1 信号通路诱导自噬,促进主动脉外膜纤维化[47]。在系统性红斑狼疮中,IL-6 处理细胞后导致自噬流明显受损,若给予托珠单抗可在一定程度上缓解自噬流受损[48]。
6 总结与展望
综上所述,促炎性细胞因子通过调节自噬在肿瘤、感染与免疫等多种疾病发展中发挥重要作用,但在AD 和PD等神经系统变性疾病中促炎性细胞因子对自噬调节的研究较少。自噬和神经炎症均在神经系统变性疾病病理机制中发挥重要作用,且两者之间存在相互调控,未来需要进一步的深入研究以了解自噬与神经炎症之间相互平衡的复杂性及潜在的分子机制,为精准调控自噬与神经炎症提供理论依据,从而找出疾病治疗潜在的靶点。
