负载型金属催化剂原子级表征及研究进展
2021-04-17杜雪丽
白 羽 满 毅 柳 颖 杜雪丽
(中国石油化工股份有限公司北京化工研究院分析研究所,北京 100013)
1 引言
随着社会的不断进步和科技的飞速发展,催化剂在人们日常生活中的地位显著提高,尤其在与人们生活息息相关的领域,例如石油冶炼[1]、有机合成[2]、环境污染的防治[3]等方面,化工产品数量和质量的提高也意味着对催化剂要求的不断提高。负载型金属催化剂在化学反应中只降低反应的活化能,不改变其在反应前后的种类和数量的属性,使得这种催化剂在化工行业中起到至关重要的作用[4],例如乙炔的选择性加氢制乙烯反应[5]、水汽转化反应[6]、催化脱氢反应[7]等。
负载型金属催化剂主要由载体和金属化合物配合而成。这种催化剂具有选择性高、活性高、稳定性高、腐蚀性小的特点,并且可以重复利用[8,9]。这些性能与催化剂结构也有很大关系。为了探究其性能和结构的关系,则需要通过多种表征手段分析。传统的表征方法目前能对不同种催化体系得到较好的剖析,例如场发射扫描电镜(SEM)可以探究催化剂的表面形貌和大小[10],X射线光电子能谱(XPS)可以探究负载催化剂表面的化学状态[11]。而对于许多依赖于表面和界面过程的催化剂来说,其原子级别结构是极其重要的。因为这些表面和界面的独特特性通常来自于它们与本体结构的尺度偏差。在非均相催化剂中,负载型金属催化剂从纳米颗粒到(亚)纳米团簇,再到单原子,其微观几何结构、表面金属原子的化学配位环境和电子结构发生了重大的变化,不仅可以显著改善催化反应活性和选择性,还能够极大地提高金属的原子利用率[12]。常用的原子级表征方法有扫描透射电子显微镜(STEM)[13]、探针分子红外光谱(FTIR)[14]等。文中将以负载型金属催化剂的表征方法为主线,综述了近年来负载型金属催化剂的原子级表征研究的进展。
2 表征方法
2.1 扫描隧道电子显微镜(STM)
STM是由诺贝尔奖获得者G. Binning和H. Rohrer根据R. Young的场发射原理的启发下创造出来的[15,16]。通过STM,使得材料晶格及原子结构被观测到,也使得加工原子尺度的新型量子器件成为可能。STM的工作原理基于量子力学中的隧道效应。在STM技术中,隧道效应产生于导体尖端和样品表面之间。具体通过使用微小的针尖在离样品表面约10埃的地方进行扫描同时加上2~3 V的电压,产生10-7~11-9A的隧道电流,测定其隧道电流就可以得到显微图像,而不用任何电子光学系统[17]。隧道电流受与应用于尖端或样品的偏差、尖端到样品的距离z、尖端和样品的形态的局部密度ρs(k, E)和其他因素的影响。因为隧道电流与尖端表面距离呈指数关系,因此表面原子的波纹可以用0.1 Å或更高的分辨率追踪[18],而固态物体原子间间距在零点几个纳米级别,因此在STM的观察下,导电物质中的原子和分子清晰可见[19]。
F. Atamny[20]等人通过研究铂金属粒子沉积在不同C材料上的表征结果来探究STM表征的局限性和潜在能力。结果表明,Pt/石墨 (HOPG)在第一个扫描循环和第二个扫描循环中同一位置的扫描图中沉积粒子有着明显差异,如图1(a)(b)所示,这表明STM技术的探针尖端可能会引起沉积沉积粒子的位移。当催化剂经过催化氧化处理和刻蚀处理后,通过STM表征,可明显观察到铂粒子沉积在石墨(HOPG)上,如图1(c)(d)所示。而当样品的负载物过于粗糙(如Pt/炭黑催化剂)则很难区分出炭黑中Pt金属粒子。虽然STM可以表征在不同条件下的催化剂局部结构信息,但是该技术经常受限于STM探针和难以区分不同化学组分的缺陷。

图1 STM图像(a).Pt/石墨催化剂模型,第一个STM扫描循环,500*500nm;(b).Pt/石墨催化剂模型,第二个STM扫描循环,500*500nm;(c).Pt/石墨催化剂通过20min500℃的加热过程后的STM图,800*800nm;(d).经过30min600℃氧化后形成的刻蚀坑的STM图,1000*1000nm;(e). Pt/石墨催化剂,120*120nm;(f).未经处理的炭黑样品,120*120nm[20]
随后人们在实际应用中对STM做了许多改进。例如在表面形貌观察方面,扫描隧道显微镜由最开始的和超真空结合系统发展到低温、反应室和其他测试仪器相结合的系统[21]。通过在实际应用中对STM的改进,使得STM可以在真空、液体、大气状态下工作;同时对样品的表面也无特殊要求;可以测量单晶、多晶、纳米相的样品。这些优越的性能特点让STM在表面科学研究方面成为一种强有力的工具。
Kwang Taeg Rim[22]等人提出了在超高真空下的Au/Fe3O4模型催化剂。为研究金原子在氧化物上的特异性位点吸附,利用扫描隧道电子显微镜观察了在受控的超高真空环境中活性物质的大小、形状、几何结构和氧化状态以及金属氧化物与金的相互作用。结果表明当Fe3O4(111)表面上的多层Au沉积被退火到500 ℃,在真空中退火15分钟后形成如图2(a)中所示,形成六边形结构(7.7×7.7×1.3 nm3), 而一些小的Au纳米颗粒(约1nm)似乎紧密地吸附在氧化物载体上,如图2(b)所示。通过STM的观察可发现Au原子沿与Fe原子间距相同的2×2表面的3倍中空位点被吸附,如图2(c)所示。

图2 Au/Fe3O4的STM图(a). 500°C退火后的Au/Fe3O4显示为一个多切面的金纳米结构,其尺寸约为7.7 nm×7.7 nm×1.3 nm;(b) .STM图像(16.5 nm×16.5 nm)显示了在原子分辨的Fe3O4(111)衬底上出现的小的Au纳米颗粒(约1 nm);(c).STM图像中黑点表示Au原子沿与Fe原子间距相同的2×2表面的3倍中空位点被吸附[22]
2.2 扫描透射电镜(STEM)
除了扫描隧道电子显微镜外,扫描透射电子显微技术也是目前应用最广泛的电子显微表征手段之一。相较于传统的高分辨相位衬度成像技术,STEM可以提供更高分辨率、对化学成分更加敏感的图像。因此可广泛应用于原子级别的微观形貌分析和成分研究。特别是用原子尺度电子探针扫描样品得到的高角环形暗场像(HAADF-STEM,Z衬度像)为非相干高分辨像,其中的亮点能真实反映原子和原子对[23]。
扫描透射成像是利用会聚电子束在样品上扫描形成的。场发射电子枪发射相干电子,经过会聚镜、物镜前场及光阑,从而会聚成原子尺度的电子束斑。束斑在样品上逐个点进行光栅扫描的同时,样品下方的环形探测器同步接收高角度散射电子,从而形成扫描透射像,如图3(a)所示[24]。入射电子束和样品间发生相互作用时,会使电子发生弹性散射和非弹性散射导致入射电子能量方向发生改变,因此在样品下方不同位置会接收到不同信号。如图3(b)所示,在θ1范围内,此时可以得到高角度非相干散射电子(HAADF,Z称度像)[24]。
配备球差校正技术的透射电镜可改善像差带来的像分辨率低和信噪比差的影响,使电镜获得更小的电子束斑(小于或者等于0.2 nm)和更高的束斑电流强度,使Z衬度像分辨率进一步提高,电镜分辨率达到亚埃尺寸,可以获得单个原子的成像[25]。由于STEM 图像的亮度与原子序数的平方成正比,即原子序数越大的原子在STEM 图像中就越亮[26],因此用STEM可直接观测到载体上活性金属原子的分布情况。
近几年,球差扫描电镜在原子尺度上深入了解材料学表征中上发挥了重要的作用。以Au/TiO2催化剂为例[27],当Au被金属氧化物(如纳米颗粒)负载时,金会显示出较高的催化活性。大量实验证实了金颗粒与氧化物载体的周界界面对催化活性起着重要作用。因此,阐明Au纳米颗粒和TiO2界面的原子和电子结构具有重要意义。
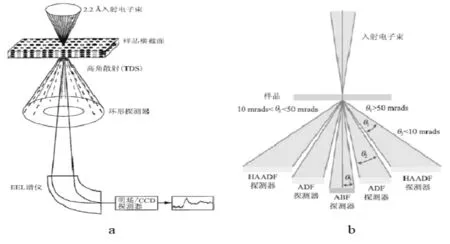
图3 STEM成像原理图(a).STEM原理图;(b).STEM中探测器分布图[24]
同时STEM技术在没有任何金属簇或NPs的情况下,可确认单个金属原子的存在以及单个金属原子相对于载体表面结构的位置,从而确定单个金属原子的空间分布[28]。图4显示了在ZnO纳米线的{10?傆b10}纳米面修饰的Au单个原子(图4a)和Pt1/Fe2O3单原子催化剂中的Pt原子(图4b)的球差校正高角度环形暗场扫描电子显微镜(HAADF-STEM)图像[29]。单个的Au和Pt原子(亮点)在极佳的图像对比度下清晰可见。当单一金属原子嵌入到二维的载体材料的表面或位于载体的内部区域,在良好的条件下EELS和EDS技术都可以用来识别单个原子的原子种类并提供被探测原子的氧化态信息。

图4 HAADF-STEM图像(a).用单个Au原子分散的ZnO纳米线; (b).Pt1/Fe2O3单原子催化剂[29].
T.Akita等人[30]用HAADF-STEM观察了金颗粒与真空沉积形成的TiO2(110)衬底之间的界面。如图5所示,入射电子束沿TiO2[001]带轴放置,可以观察到沿[110]带轴平行于电子束的金粒子的晶格条纹。在图5(a)中,TiO2(110)表面的金颗粒(111)平面倾斜15度。图5(b)中Au(111)平面与TiO2(110)平行。图5(c)中Au(200)平面与TiO2(110)平行。大部分的金颗粒都倾向于负载在TiO2基体的表面台阶位置,少量金粒子Au[1~10]轴与TiO2[001]轴平行。HAADF-STEM方法尽管很难看到像O原子这样的轻原子,但具有直接求解原子构型,无需考虑离焦值和样品厚度的图像模拟的优点,因此它是最为广泛的原子级表征方法之一。
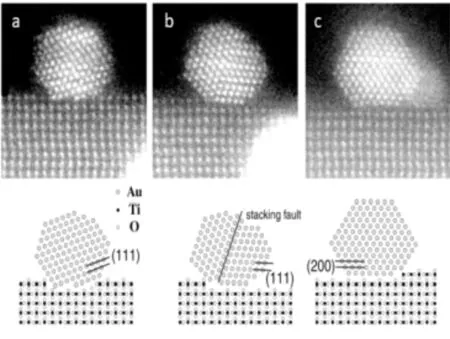
图5 TiO2 (110)上金颗粒的HAADF-STEM图像[30]
2.3 探针分子红外光谱(FTIR)
尽管STEM是负载型催化剂一种常见的表征方法,但是STEM无法提供关于单个金属原子的局部环境的细节,除非负载原子的轮廓是清晰的。如果将STEM与其他表征方法结合,可分析整个大样本中单原子存在的均匀性,并且对负载的单原子局部环境的深入了解都是非常有帮助的。
在众多催化剂表征方法中,原位红外光谱表征技术在加氢催化剂中应用十分广泛[31],可以在分子或原子水平上研究催化剂活性相的信息。因为它可以直接监测吸附(探针)分子与固体金属或载体表面之间的相互作用[32]。该方法利用分子探针与固体表面的相互作用来提取有关吸附探针分子的物质性质的信息。通过监测探测模式的振动频率和强度的变化以及适当的校准推断出活动中心的性质。时间和温度分辨的傅里叶变换红外光谱(FTIR)可以用来检测催化反应中的中间体[33]。同时也是一种强大的特定位点表征技术,这种技术不仅可用于确定和量化催化剂样品中负载物的浓度,对于负载物种的局部几何结构、稳定性、活性和均匀性提供有效数据。在催化剂的红外光谱表征中,探针分子的选择对于确保被吸附分子的频率或能带形貌对吸附位点的各种特性做出响应,从而进行位点特异性分析是非常重要的。CO探针分子在过渡金属上具有良好的配位性能,已成为应用于金属催化剂研究的常用的探针分子。通过研究探针分子在催化剂活性金属上的吸附,可以研究催化剂不同吸附位对应的特征峰,这为研究催化剂活性位提供了信息。
吴宇航[34]等人通过CO探针红外光谱法探究了CoMo/Al2O3-SiO2催化剂催化中心结构和作用过程的相关信息。并得到如图6所示的结果。按照Eisehens等[35]根据已知结构的金属羰基化合物的红外光谱图所总结出的规律,υco>2 000cm-1归属为线式CO吸附态, υco<2 000 cm-1归属为桥式吸附态。图6(a)所示可以看出,探针分子在硫化态CoMo/Al2O3-SiO2催化剂上的吸附主要线性吸附。如图6所示,硫化温度在300~550℃范围内时,该催化剂的MoS(2110cm-1)和CoMoS(2070cm-1)活性相均同时存在。但在硫化温度较低(300℃)时,MoS相占据比例大于CoMoS相;随着硫化温度增加到500℃,催化剂活性相结构发生变化,CoMoS相占据比例大于MoS 相,表明部分MoS相转变为CoMoS相,增加了活性点位。
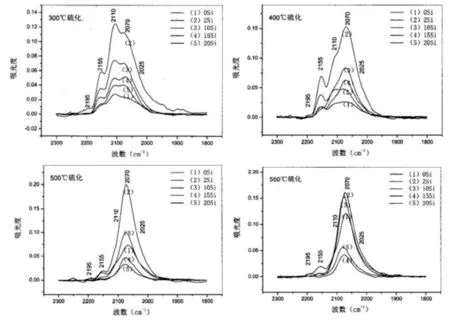
图6 不同硫化温度下CoMo/Al2O3-SiO2催化剂吸附CO的红外谱图(a).300℃;(b)400℃;(c).500℃;(d).550℃硫化CoMo/Al2O3-SiO2催化剂吸附CO的红外光谱[33]
Matsubu等人[36]以CO作为探针分子,将新合成的4% Rh/TiO2催化剂上300 K下进行CO吸附。所得红外光谱以Kubelka-Munk(KM)单位显示,并以对称的偕取二羰基峰(2097 cm?傆b1)高度进行归一化,以便进行比较。通过图7(a),作者将-2097和-2028cm?傆b1的峰指定为Rh(CO)2偕取二羰基峰的对称和不对称延伸,它们只存在于TiO2载体的独立Rh原子点位。不同Rh负载的光谱清楚地显示了线性CO吸附在孤立的Rh单原子位点上的峰值强度变化(图7b),并且不同类型的点位的含量也可以被量化。这些数据表明,在Rh负载极低的情况下,Rh主要以孤立的单个原子的形式存在。随着Rh负荷水平的增加,则形成Rh簇或纳米粒子。
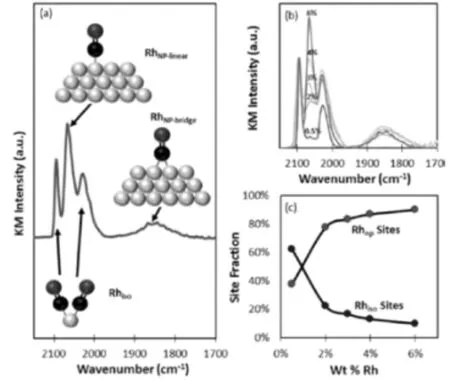
图7 4% Rh/TiO2催化剂上300 K下进行CO吸附(a). 300 K吸附在4% wt % Rh/TiO2上的CO饱和层获得的DRIFT谱; (b).CO在5种重量负载(wt %) Rh/ TiO2催化剂上的DRIFT谱; (c).孤立的的Rhiso和基于纳米颗粒的Rh位点的位点含量(%)[36]
2.4 X射线吸收光谱(XAS)
X射线吸收光谱是由吸收两侧的小峰和波动构成,由吸收原子周围的近程结构决定,可提供小范围内原子簇结构的信息[37],数据通过傅里叶变换得到径向结构矢量图,横坐标表示原子间距离(R),纵坐标则表示波的矢量(k)。通过这些数据可得到活性金属原子的电子结构、配位环境、金属-载体相互作用等信息[38]。
Zhang等[39]对Pt/Ni(OH)X催化剂进行了XAF表征,发现Pt通过与O原子和Ni原子形成Pt-O键、Pt-Ni键与Ni(OH)X载体形成金属-载体相互作用,从而说明Pt主要是以孤立原子的形式分散于Ni(OH)X载体。Isao Ogino[40]利用XAS技术表征以Sn-β-沸石催化剂为典型的金属酶类催化剂其活性点位的结构信息。由于类金属酶类催化剂具有高均一性结构点位,通过EXAFS数据其R值可达到5 Å,从而准确测定Sn原子的位置(图8)。
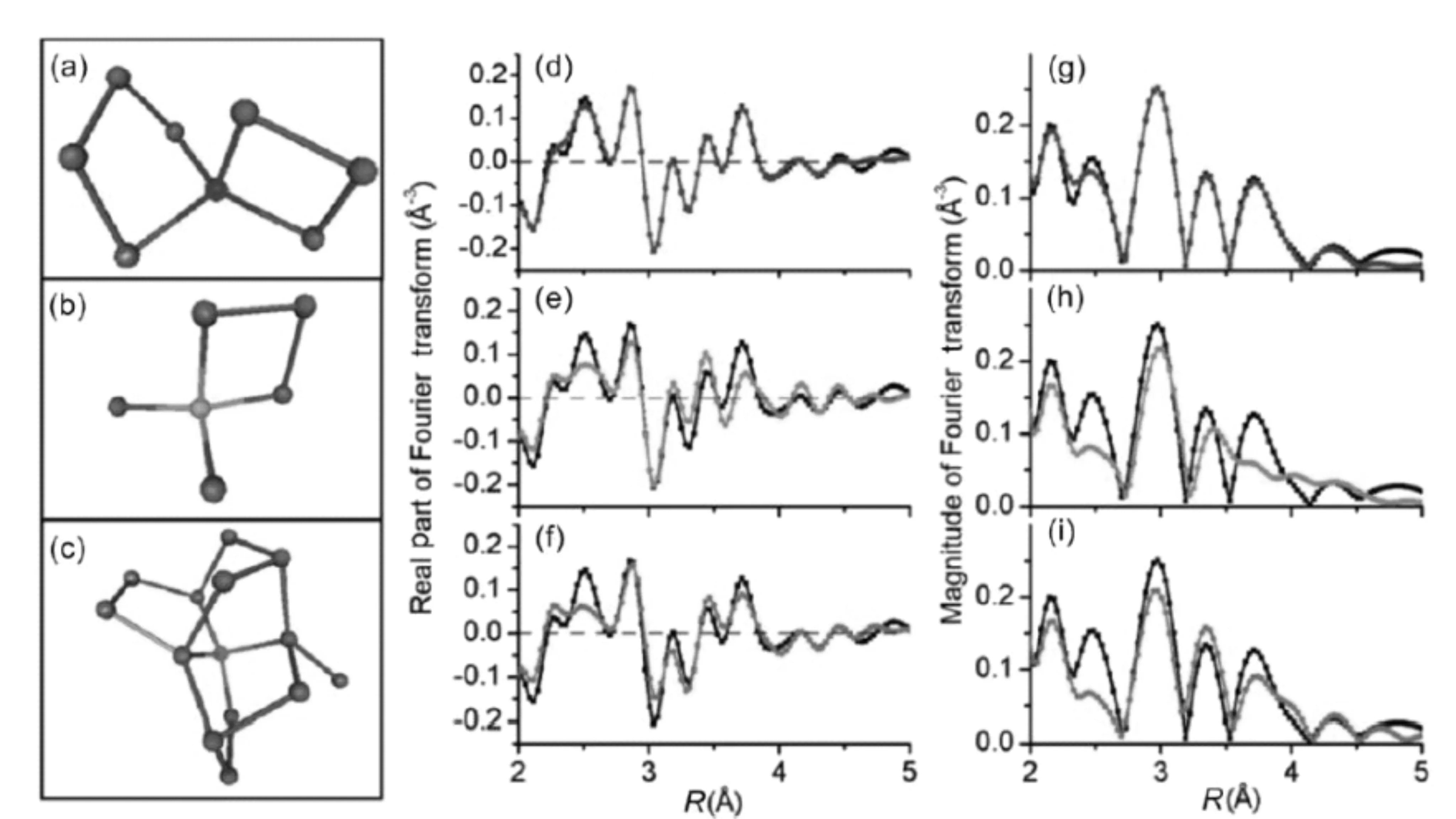
图8 Sn-β沸石催化剂的EXAFS数据图 [39]
周功兵等人[41]通过对在最优条件下Ru-Pd/ZrO2-0.2和Ru-Pt/ZrO2-0.15催化剂(Pd、Pt与Ru的摩尔比分别为0.2和0.15)的微观结构进行了表征,通过EXAFS得到了Ru K边k2χ(k)数据的傅里叶变换径向分布函数,向分布峰的位置表示吸收原子和近邻原子间的距离。通过XANES谱得到催化剂能量谱。从图9(a) (b)中可以看出两种催化剂与Ru箔和Pt粉末的Ru K及Pt LⅢ边能量相近 说明催化剂中的Ru和Pt为金属态。Ru-Pt/ZrO2-0.15催化剂在在1.88 Å出现Ru-B配位峰,在2.36 Å处出现Ru-Ru的第一近邻配位峰, 说明金属纳米粒子具有非晶态结构。同时催化剂的径向分布峰明显弱于Ru箔,表明催化剂上的Ru配位不饱和。
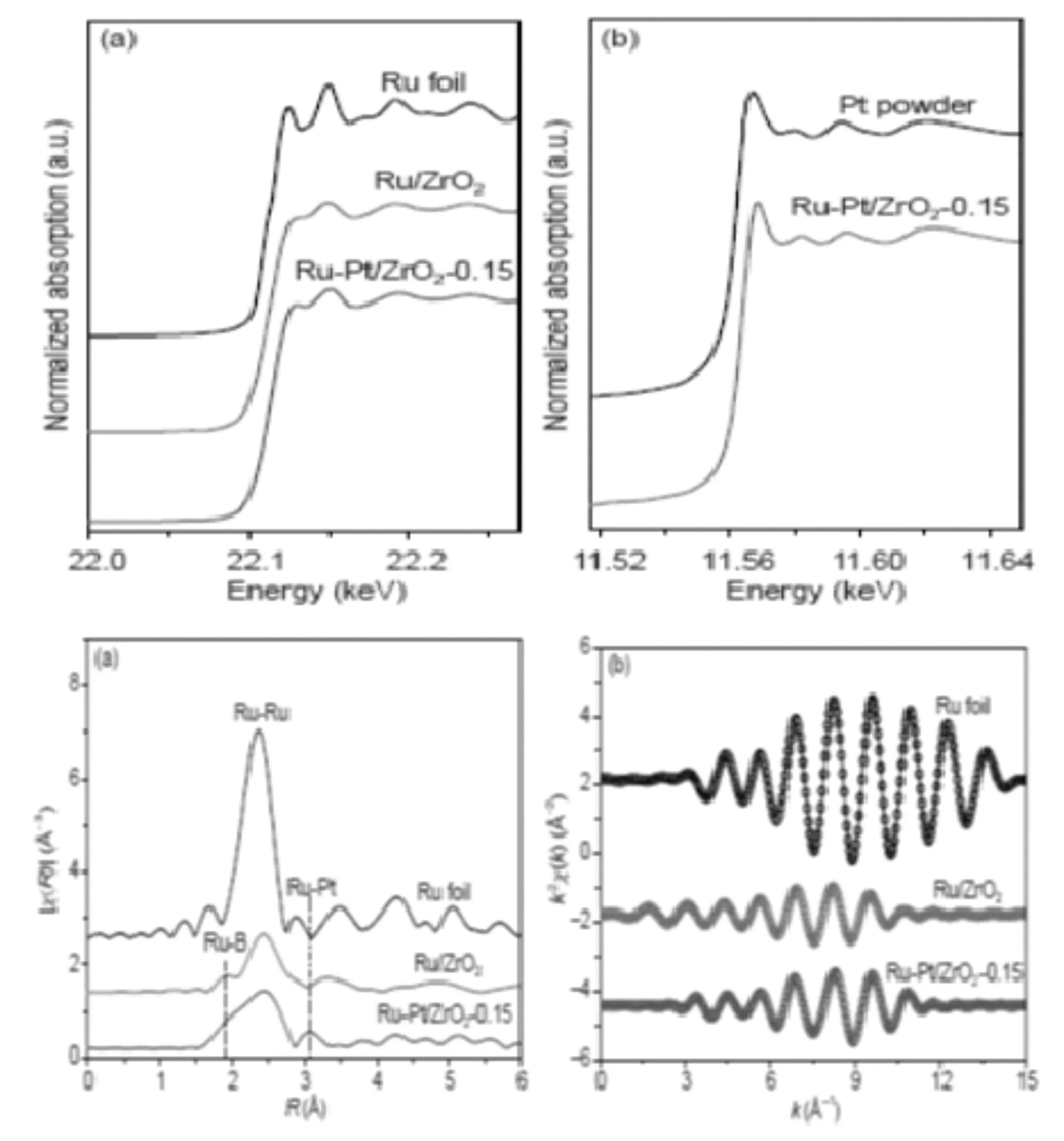
图9 Ru/ZrO2、Ru-Pt/ZrO2-0.15催化剂及Ru箔、Pt粉末标样归一化后的普图(a).Ru K边;(b).Pt LIII边XANES谱; (c).Ru/ZrO2、Ru-Pt/ZrO2-0.15催化剂及Ru箔标样Ru K边k2χ(k)数据的傅里叶变换径向分布函数(相移未校正); (d) .Ru/ZrO2、Ru-Pt/ZrO2-0.15催化剂及Ru箔标样Ru K边k2χ(k)实验数据(O)及拟合曲线(—)[41].
3 结论
简要介绍了负载型金属催化剂的一系列原子级表征方法。为了解负载型金属催化剂在非均相反应中表面金属原子的化学配位环境和电子结构发生的变化,广泛应用了有扫描隧道电子显微镜(STM)、扫描透射电子显微镜(STEM)、探针分子红外光谱技术(FTIR)、X射线吸收光谱(XAS)等分析表征技术。通过这些原子级别的分析表征手段显著改善催化反应活性和选择性,还能够极大地提高金属的原子利用率。同时根据催化剂的实际情况,采用TPD、XPS、XRD等辅助性手段进行表征,通过多种表征手段的融合,可获得催化剂的局部信息和整体平均信息,最终实现催化剂的实际工业价值。
