Bacterial and archaeal communities in deep sea waters near the Ninetyeast Ridge in Indian Ocean*
2021-04-14PingGAOLingyunQUGuangxunDUQinshengWEIXueleiZHANGGuangYANG
Ping GAO , Lingyun QU,**, Guangxun DU , Qinsheng WEI ,3, Xuelei ZHANG,Guang YANG
1 Key Laboratory of Marine Eco-Environmental Science and Technology, First Institute of Oceanography, Ministry of Natural Resources (MNR), Qingdao 266061, China
2 Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266237, China
3 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology(Qingdao), Qingdao 266237, China
Abstract Depth-dependent distribution patterns of bacterial and archaeal communities in deep sea water column around the Ninetyeast Ridge in the Indian Ocean were investigated using 16S rRNA gene profiling.Sampling was conducted at the northern Ninetyeast Ridge (1°59.89′N-9°59.70′S, 87°58.90′E-88°00.03′E)from September to November 2016 where samples were collected from the bathyal (1 000 m) to bathypelagic depths (>4 000 m) in four different stations. A total of 1 565 405 clean data falling into 6 712 bacterial OTUs and 1 452 727 clean data falling into 806 archaeal OTUs based on 97% similarity level were analyzed. Most of the bacterial 16S rRNA gene sequences were affi liated with Gammaproteobacteria,followed by Alphaproteobacteria and Bacteroidia. The archaeal 16S rRNA gene sequences mostly affi liated to Nitrososphaeria (Thaumarchaeota) dominated with relative abundances ranging from 52.68% to 97.2%,followed by Thermoplasmata (Euryarchaeota). Vertical partitioning of bacterial and archaeal communities among diff erent water layers was observed. Canonical correspondence analysis (CCA) and Spearman’s correlations revealed that depth ( P=0.003), dissolved oxygen ( P=0.019), and nitrite ( P=0.033) were the main environmental factors aff ecting bacterial community structure at genus level in the Ninetyeast Ridge.On the other hand, the first two CCA axes accounted for 74.4% of the explained total variance, it seems that the archaeal communities at genus level were heavily influenced by the environmental variables including depth, dissolved oxygen (DO), nitrite, salinity, phosphate, ammonia, nitrate, and silicate, but none of them exhibited any significant correlation on the structuring ( P >0.1).
Keyword: deep sea water; the Ninetyeast Ridge; 16S rRNA gene; bacteria; archaea
1 INTRODUCTION
Microbes have been shown to be indispensable components of marine ecosystems, from the surface waters (Bouvier and de Giorgio, 2002; Cunliff e et al.,2011) to the sediments in estuaries (Guo et al., 2018),and even in the extreme environments including hydrothermal vents (Zhang et al., 2016), ice sheets in polar regions (Christner et al., 2014), and the bottom of the Mariana Trench (Liu et al., 2019). Marine microbes play important roles in various biological and ecological processes, including but not limited to organic matter decomposition (Feng et al., 2009),carbon cycling through bacterial biomass production and remineralization of dissolved organic matter via respiration (McArthur, 2013; Kirchman, 2016),nitrogen fixation (Barlett and Leff , 2010), nitrogen removal (Qian et al., 2018), sulphate reduction (Guo et al., 2018) among others. Currently, knowledge on microbes in deep seas or water depths below 200 m where the sunlight required for photosynthesis is absent (Terahara et al., 2016) is relatively scarce.
The Indian Ocean (IO) is characterized by its complex physical dynamics and variable systems including the presence of several ridges. Among these ridges, the Ninetyeast Ridge that runs S-N (slightly to the East) orientation and extends along 90°E more than 5 600 km from 34°S to 17°N (Krishna et al.,1999) is the longest aseismic (not associated with active faulting) ridge in the world. It also separates the Central Indian Basin from the Cocos and West Australian Basins (Krishna et al., 1999; Michael and Krishna, 2011). The north of the Ninetyeast Ridge where is buried by sediments coming from the Bengal Fan, which is deeper than 4 000 m. Recently, microbial communities in the sediments of the vents in the Southwest Indian Ridge has been described (Zhang et al., 2016). However, despite the unique tectonic,topographic, bathymetric, and hydrographic features of the Ninetyeast Ridge, the composition, diversity,and distribution of bacteria and Archaea in horizontal and vertical gradients approaching the Ninetyeast Ridge remain little understood.
Many factors influence the distribution and ultimately the structuring of bacterial communities in water or sediments. For example, the previous study reported that dissolved oxygen was the most important environmental factor determining the distribution of anammox bacteria in the deep water samples (500-2 000 m) collected from the Eastern Indian Ocean(Qian et al., 2018). It is also reported that SO42ˉ,salinity, and total phosphorus were the vital environmental factors aff ecting bacterial community structuring in the intertidal sediments of Changjiang(Yangtze) River estuary (Guo et al., 2018). In other bacterial communities, major drivers include salinity for halophilic species (Bouvier and de Giorgio, 2002),nitrogen level for denitrifiers (Zheng et al., 2014), and sulphate for sulphate-reducing prokaryotes (Guo et al., 2018). Deep-sea environments exhibit complex dynamic habitats that are characterized by steep thermal and chemical gradients. However, it remains unclear on how bacterial and archaeal communities respond diff erently to the changes in environmental conditions along the vertical gradient from bathyal to abyssopelagic depths. Ifit occurs, what are the major factors regulating vertical variations in bacterial and archaeal communities around the Ninetyeast Ridge?
Generally, most marine microbes are uncultivable,and with the advent of molecular methods and highthroughput sequencing, more and more discoveries have been made in exploring the biodiversity and biogeography of deep-sea microorganisms. In this study, samples were collected from stations along 88°E (longitude) dissecting the Ninetyeast Ridge,where water samples were collected at diff erent depths from 1 000 m to depths more than 4 000 m(bottom layer). Specifically, thus study aims to (1)assess the diversity and taxonomic composition of bacterial and archaeal assemblages from bathyal to the abyssopelagic zone, and (2) understand the major environmental parameters potentially driving the distribution and diversity of bacterial and archaeal communities. To our knowledge, this is the first study that explored the horizontal and vertical distributions of bacterial and archaeal communities in deep sea waters near the Ninetyeast Ridge in the Indian Ocean.
2 MATERIAL AND METHOD
2.1 Sample sites and collection
Sampling was carried out from September to November 2016 during a cruise to the East Indian Ocean onboard the R/V Xiang Yang Hong 1. To further understand bacterial and archaeal communities and their responses to environmental factors in deep waters, seawater samples were collected in four stations (S703, S708, S711, and S717) from diff erent depths were selected (Fig.1) along the Ninetyeast Ridge. Immediately after seawater collection, 2-L seawater from each depth was filtered through 0.22-μm pore size membrane (Waterman, France). The filter membranes were immediately frozen at -80 °C until nucleic acid extraction.
2.2 Physicochemical parameters
For each sample, in situ measurements of depth,temperature ( T), salinity, and dissolved oxygen (DO)were recorded from different sensors fitted onto the CTD (Sea-Bird Electronics, USA) rosette system.The measurement methods for ammonia, nitrate,nitrite, phosphate, silicate followed those recommended by Parsons et al. (1984). Nitrate and nitrite were detected by the standard pink azo dye method and ammonia using the hypobromate oxidation-pink azo dye method. Meanwhile,phosphate and silicate were detected using the standard molybdenum blue method (Parsons et al.,1984).
Fig.1 Sampling sites approximate to the Ninetyeast Ridge in the Eastern Indian Ocean
2.3 DNA extraction and PCR amplification
Total genomic DNA of the water samples was extracted using the water DNA Kit (OMEGA BioTek Inc., Doraville, GA, USA). DNA concentration and purity were checked with 1% agarose gels. DNA were then diluted to 1 ng/μL using sterile water. The primer set 341F (5′-CCTAYGGGRBGCASCAG-3′)and 806R (5′-GGACTACNNGGGTATCTAAT-3′)was used to amplify the V3-V4 region of bacterial 16S rRNA gene (Michelsen et al., 2013). At the same time, the primer set Arch519F(5′-CAGCCGCCGCGGTAA-3′) and Arch915R(5′-GTGCTCCCCCGCCAATTCCT-3′) was used for the amplification of the V4-V5 region of archaeal 16S rRNA gene (Hugoni et al., 2015).
2.4 Library preparation and 16S rRNA gene sequencing
Sequencing libraries were produced by Ion Plus Fragment Library Kit 48 RXNS (Thermo Scientific)and the library quality was assessed on a Qubit@2.0 Fluorometer (Thermo Scientific). Then, the prepared libraries were sequenced on an Ion S5TMXL platform and 400 bp/600 bp single-end reads were generated at Novogene (Beijing, China).
2.5 DNA sequence processing and analysis
Single-end reads were divided into diff erent samples based on the unique barcodes and truncated by cutting off the barcode and primer sequence. Then quality filtering on the raw reads were carried out by specific filtering instructions to obtain clean reads following the pipeline suggested in Cutadapt (Martin,2011). Afterwards, chimeric sequences were detected and removed using UCHIME algorithm (Edgar et al.,2011; Haas et al., 2011).Sequence analyses were implemented using Uparse software (Edgar, 2013). Sequences in ≥97% similarity were assigned to the same OTUs and then representative sequences for each OTU were picked for further annotation using the Silva Database(https://www.arb-silva.de/) (Quast et al., 2012).
2.6 Diversity and statistical analyses
Alpha diversity was calculated in Quantitative Insights into Microbial Ecology (QIIME)(Version1.7.0) and displayed with R software (Version 2.15.3). It was used to determine species diversity within samples using diff erent indices, including Chao1 (Chao, 1984), Abundance-based Coverage Estimator (ACE) (Chao et al., 1993), Shannon and Good’s coverage. Chao1 and ACE correspond to the richness of the community while Shannon index estimated the diversity of microbial species. In order to explain the scope of bacterial and archaeal diversity,Good’s coverage (C) was calculated according to Li’s instructions (Li et al., 2014).
Unweighted Pair-group Method with Arithmetic Means (UPGMA) (Li and Xu, 2007) was used for the visualization of beta diversity on the basis of Binary-Jaccard distance through the QIIME software (Version 1.9.1). Also, Non-metric multi-dimensional scaling(NMDS) analysis (Kruskal, 1964) was performed to visualize Bray-Curtis distances between and among microbial communities of the four sampling stations with R package vegan.
The correlation between microbial richness,biodiversity, and environmental factors was determined by Spearman’s rank correlation analysis.In addition, canonical correspondence analysis(CCA) (Sheik et al., 2012) was performed by combining correspondence analysis with multiple regression analysis to assess the correlations between microbial community patterns and environmental factors using R package vegan. In Spearman’s rank correlation analysis, the correlation coeffi cient between species and environmental factors was calculated by the “corr.test” function of the psych package in R, and its significance is tested, then the pheatmap function in the pheatmap package is used for visualization. differences were considered significant at P <0.05.
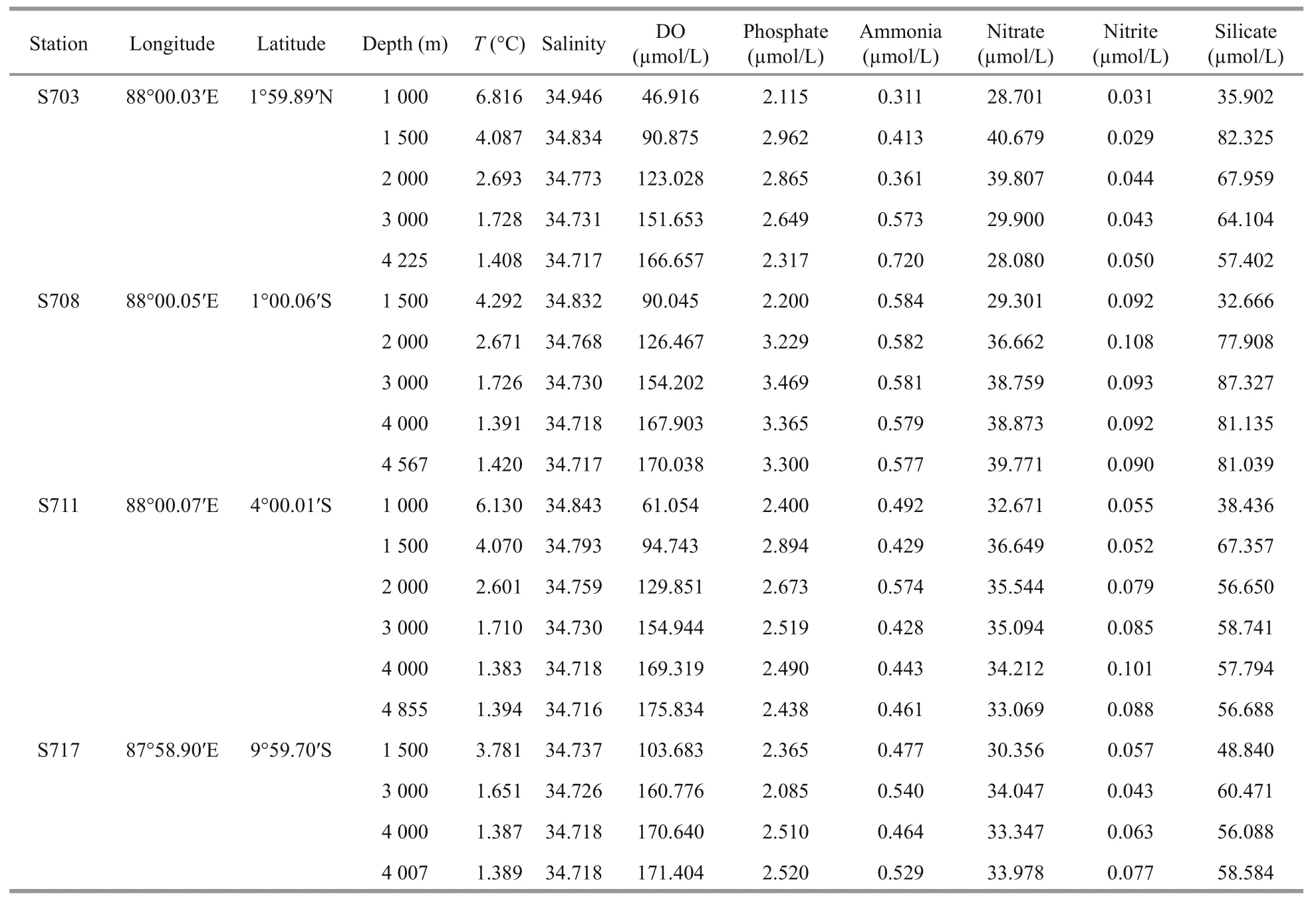
Table 1 Sampling sites description and physico-chemical characteristics
3 RESULT
3.1 Physico-chemical parameters
Summary of the environmental parameters (i.e.,depth, temperature, salinity, DO, phosphate, ammonia,nitrate, nitrite, and silicate) measured during the expedition in the Ninetyeast Ridge are presented in Table 1. Notably, temperature and salinity gradually decreased with depth in all the stations. In contrast,DO showed the opposite trend compared with temperature and salinity profiles (Fig.2). In addition,for all inorganic nutrients, their concentrations generally slightly increased before decreasing in each station. Specifically, phosphate, ammonia, nitrate, and nitrite in station S708 were detected higher at >2 000 m than its corresponding water layers in other stations(Fig.S1). These indicate strong heterogeneity in the physico-chemical conditions among the stations.
3.2 Diversity of bacterial and archaeal communities
In total, 1 644 299 and 1 538 196 raw sequences for bacteria and Archaea were respectively generated from the 20 water samples and there were deposited in the NCBI Sequence Read Archive under the accession numbers SRR10418076 and SRR10418164,respectively. After quality filtering, only 1 565 405 and 1 452 727 high-quality sequences were retained for bacteria and Archaea, respectively. Both rarefaction curves based on OTUs for bacterial and archaeal species were asymptotic, suggesting that the sampling depths were suffi cient to capture overall microbial diversities in all the samples (Fig.S2).
A total of 6 712 OTUs for bacteria and 806 OTUs for Archaea were obtained from the 20 water samples.Venn diagram showed 38 bacterial OTUs were common among the samples, making up 8.92%-15.02% of the sequences of each library (Fig.3a). The samples from stations S703, S708, S711, and S717 shared 105, 110, 63, and 103 OTUs, respectively,comprising 5.73%, 6.61%, 3.23%, and 8.15% of the sequences in each of the station (Fig.S3a). Out of 512 archaeal OTUs, only seven were common among all samples, covering 12.50%-41.18% of sequences from individual samples (Fig.3b). In addition, 11, 8,8, and 9 archaeal OTUs were common for station S703, S708, S711, and S717, respectively, contributing to around 4.82%, 3.51%, 3.60%, and 7.03% of the sequences in each station (Fig.S3b).
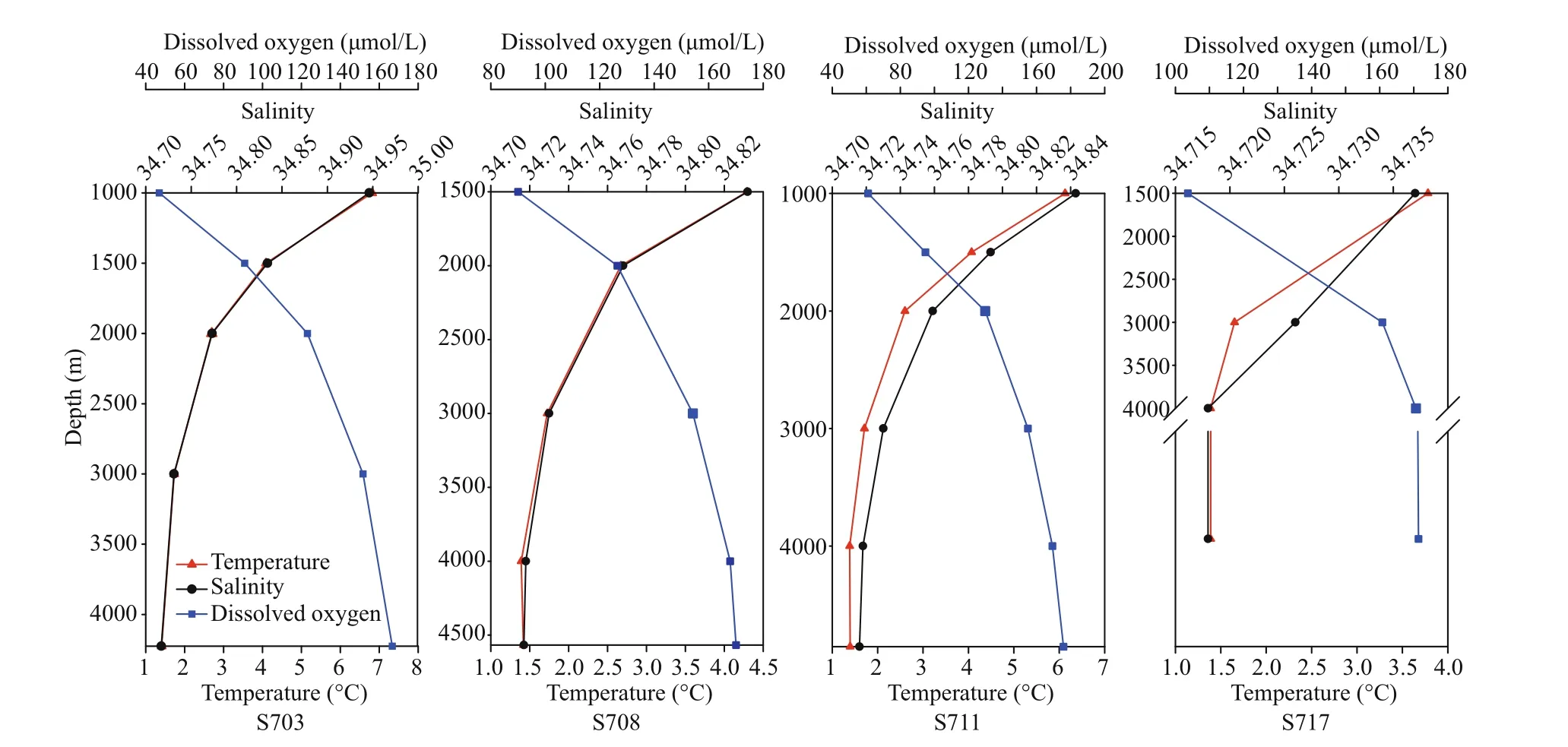
Fig.2 Vertical variations of physicochemical parameters including salinity, temperature and DO in the water column in diff erent stations (S703, S708, S711, S717) approximate the Ninetyeast Ridge in Indian Ocean
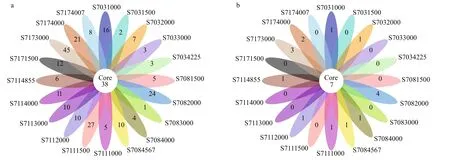
Fig.3 Flower diagrams of the unique and shared OTUs for bacterial (a) and archaeal (b) diversity
The diversity and richness of the microbial communities in water column were characterized by observed species, Shannon and Chao1-ACE indices(Table 2) and these generally decrease as depths increase in each station. The highest diversity of bacteria was observed in the depth 1 500 m of S711,followed by the 1 500 m from station S717, while the lowest bacterial diversity was in the bottom depth of station S711, followed by station S703 in the bottom layer (Table 2). The highest community richness for bacteria was found at 1 500 m depth of S717 while the lowest richness from 4 000 m of the same station,followed by the depth 4 000 m of S711.
Interestingly, for Archaea, the highest diversity was observed at 2 000 m of S711 and the highest richness at 3 000 m in S708. Similar to bacteria,lowest archaeal diversity and richness were both observed at ≥4 000 m. However, compared to bacteria,the richness of Archaea were significantly lower.
3.3 Similarity analysis among the bacterial and archaeal communities
UPGMA analysis revealed clear patterns of horizontal partitioning, which segregated all the samples into two significant clusters. The first cluster mainly consisted of bacterial communities residing in the stations of S703 and S708, and the second cluster comprising those from the stations of S711 and S717 except of the depth 1 000 m from S711 (Fig.4a).Interestingly, no such horizontal differences seen in archaeal community but there was apparently vertical partitioning in archaeal clone libraries (Fig.4b).
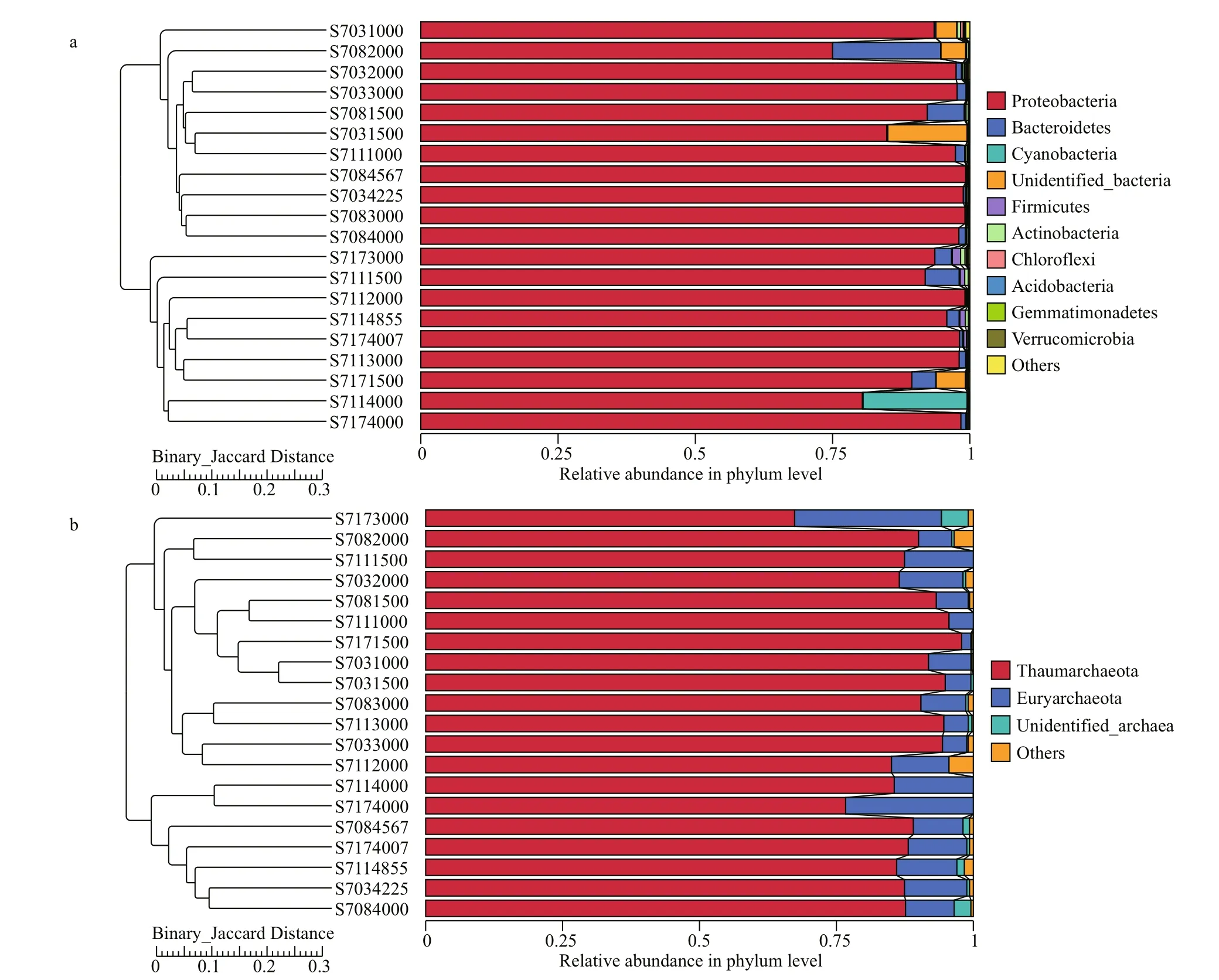
Fig.4 UPGMA analysis of bacterial (a) and archaeal (b) communities in the four sampling stations based on Binary-Jaccard Distance
Non-metric multidimensional scaling (NMDS)ordination was also employed to decipher latitudinal location and vertical similarities among bacterial and archaeal communities (Fig.5). The stress value for bacteria and Archaea is 0.066 and 0.133 respectively,suggesting a good fit of data points in hierarchical clustering analysis. As illustrated by NMDS for bacteria (Fig.5a), stations S703 and S708 were remarkably separated along the X-axis from stations S711 and S717. Thereinto, samples from S703 except of S7031500 formed a single cluster, while samples from S708 grouped anther cluster. In addition, the depths deeper than 4 000 m in stations S711 and S717 appeared to show a similar bacterial community structure as they assembled in one plot, while the depths <4 000 m did not group together. Obviously,the geographical ordering of samples showed distinguishable dissimilarities of bacterial community composition in terms oflatitude.
For Archaea (Fig.5b), samples belonging to diff erent water layers clustered respectively,suggesting there were great heterogeneities among species composition across vertical gradients of water column. It is interesting to note that no apparent grouping was observed horizontally, which implies Archaea distributed in the tested water column did not vary greatly in a smaller latitudinal scale. All these profiles indicated patterns similar to those seen from UPGMA analysis (Fig.4).
3.4 Bacterial and archaeal taxonomic assignment and community composition
Classified bacterial sequences belonged to 22 phyla, 35 classes, 82 orders, 156 families,314 genera, while archaeal sequences were from 2 phyla, 5 classes, 7 orders, 8 families, and 8 genera.The most abundant bacterial taxa were affi liated to Proteobacteria, accounting for 74.99%-99.23% of the total sequences in the four stations (Fig.S4a).Proteobacteria were higher at >4 000 m than the other layers in diff erent stations, except for S711 where it was highest (99.16%) at 2 000 m. In addition, Bacteroidetes were the second dominant phylum, with 0.16%-19.68% of the sequences at the 20 samples. Moreover, Cyanobacteria were detected in most of the samples with relatively lower abundance, however, it was interesting to note that Cyanobacteria at 4 000 m of S711 reached up to 18.93%.
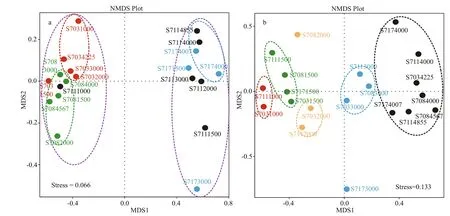
Fig.5 Nonmetric multidimensional scaling (NMDS) analysis of bacterial (a) and archaeal (b) communities
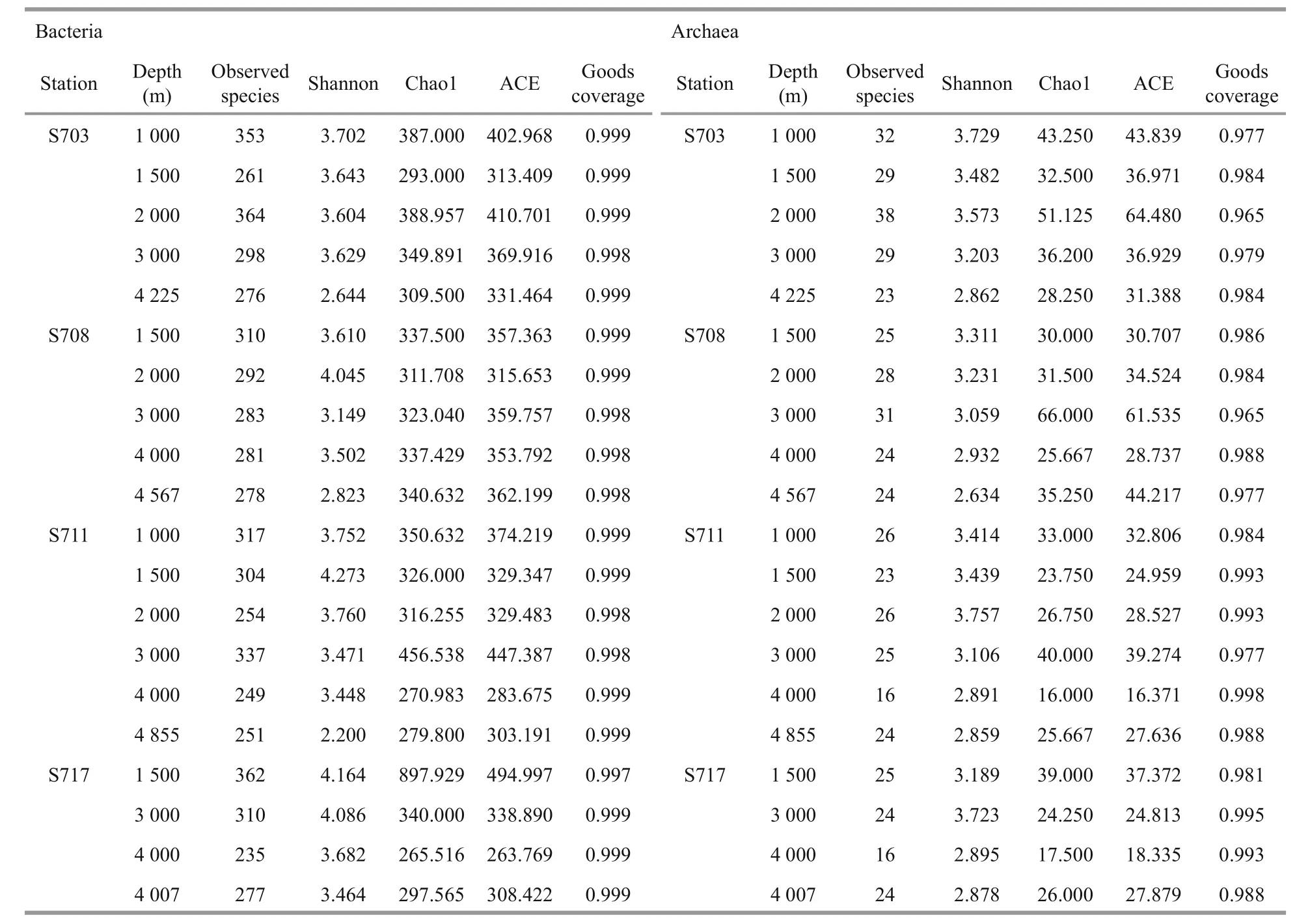
Table 2 Diversity and richness of bacteria and archaea
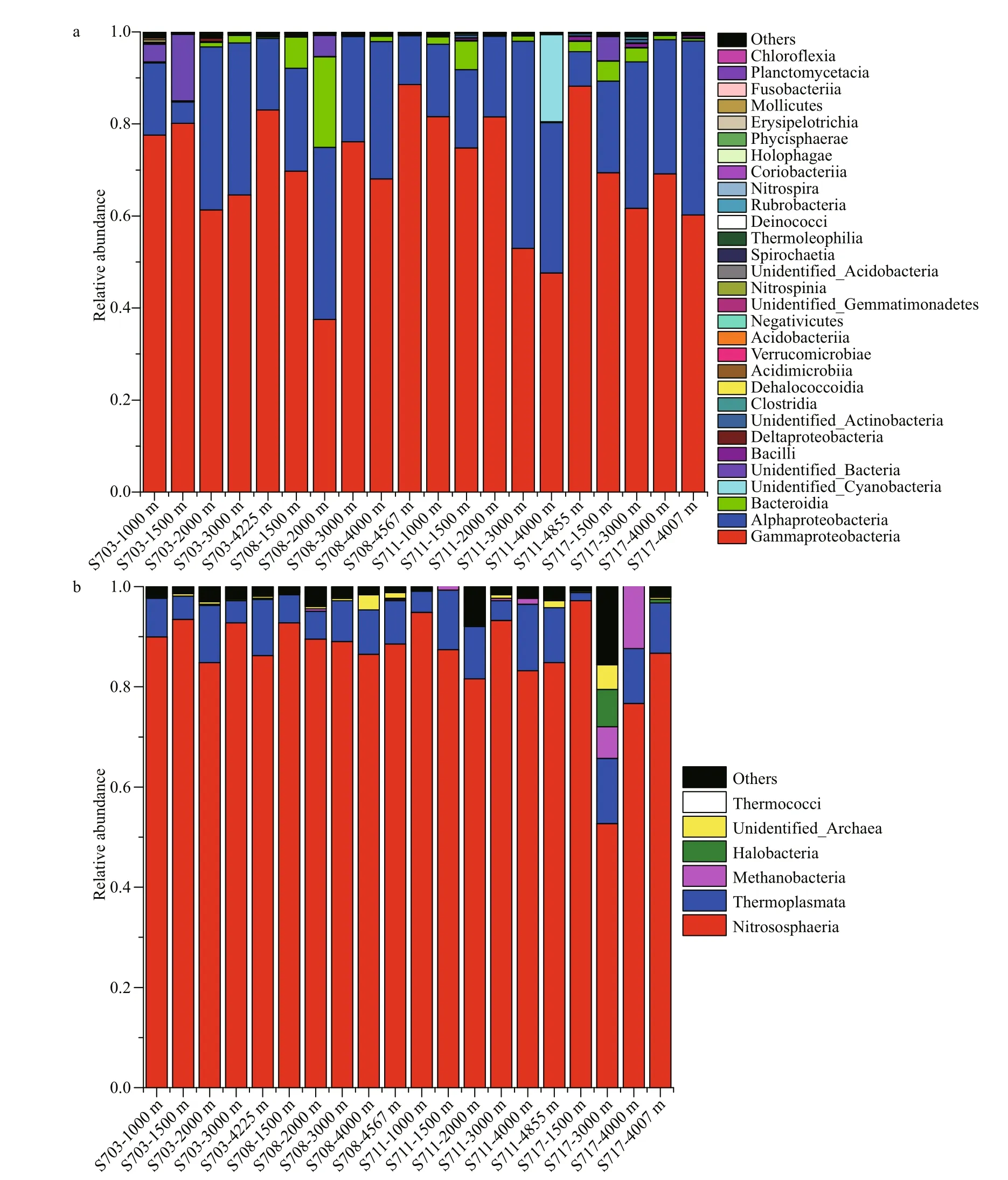
Fig.6 Histogram of relative abundance of bacterial (a) and archaeal (b) species at class level
At class level, a large proportion of sequences were taxonomically assigned to Proteobacteria,including Gammaproteobacteria (37.51%-88.55%),Alphaproteobacteria (4.62%-45.03%) and Deltaproteobacteria (0.03%-0.76%). Of that,Gammaproteobacteria consisted widely in all the samples and was distinctly rich in deeper than 4 000 m in each station, while Alphaproteobacteria was tend to be most abundant in 2 000-3 000 m and fewer in the bottom of each station. The third abundant bacterial class Bacteroidia, which subordinates to Bacteroidetes also occupied a large proportion among these taxa assemblages, but tend to be more abundant in the relative upper water layers (1 000-2 000 m) (Fig.6a,Supplementary Table S1a).
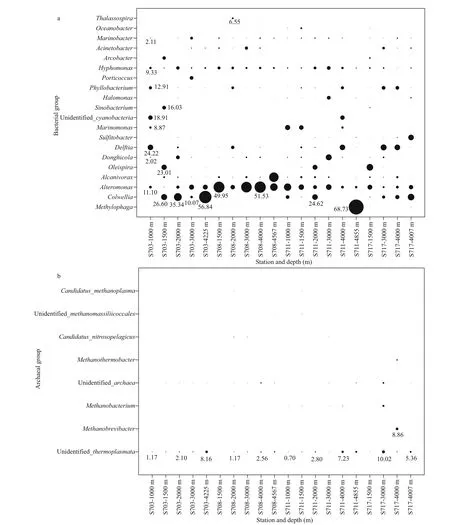
Fig.7 Depth wise distributions of percentages of bacterial (a) and archaeal (b) genus in the sea water near the Ninetyeast Ridge in Indian Ocean
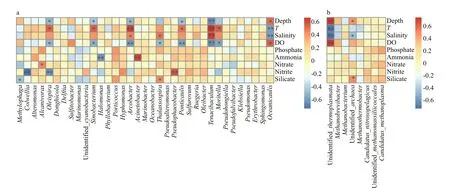
Fig.8 Heatmap summarizing Spearman’s correlations analysis between the environmental factors and alpha diversity index for bacterial (a) and archaeal (b) species at the genus level
Significant dissimilarities were observed in the relative abundances of diff erent bacterial genera in the water column. The distribution patterns of bacterial genera of top 20 (Fig.7a) indicated that overwhelming majority of sequences clustering into Methylophaga, Colwellia, Alteromonas, Alcanivorax,and Oleispira were affi liated to Gammaproteobacteria.Among the Gammaproteobacteria, Methylophaga within unclassified family demonstrated the greatest relative abundance (68.73%) in the bottom of S711 and the second abundant Colwellia consisted distinctly in all the layers of station S703 and S717,but hardly detected in S708.
As for Archaea, the most detected sequences belonged to two known phyla, while others were still unannotated and unclassified by far. In average, the majority of 88.5% sequences belonged to Thaumarchaeota; however, only 9.9% were members of Euryarchaeota, suggesting that Thaumarchaeota overwhelmingly dominated in deep seawater at the Ninetyeast Ridge (Fig.S4b). At the class level,Nitrososphaeria affi liated to Thaumarchaeota phylum were the predominant class in all the 20 samples,accounting for 52.68%-94.87%. Thermoplasmata belonging to Euryarchaeota phylum also occupied a small fraction of the detected sequences with abundance of 1.63%-13.69%, however, it seems that it is richer in bottom layers (>4 000 m) than other depths (Fig.6b, Supplementary Table S1b).
Summary of relative abundances at genus level for Archaea was presented in Fig.7b. Most of the sequences remained unannotated due to lack of possible representative sequences with complete identification by far, whereas minor sequences were clustered into eight genera, with seven affi liated to Euryarchaeota, and one Thaumarchaeota (Fig.7b).The most abundant Archaea in those samples belonged to Euryarchaeota phylum, Thermoplasmata class,followed by Methanobrevibacter and Methanobacterium, corresponding to anaerobic methanotrophic taxa in water column.
3.5 Correlation between the bacterial and archaeal communities and environmental factors
Spearman correlation analysis was carried out to study the linkage between microbial community diversity and environmental factors. It revealed that Shannon index based on the bacterial community was positively correlated with T (Spearman R=0.57,P <0.01) and salinity ( R=0.66, P <0.01) but negatively with depth ( R=-0.74, P <0.01) and DO ( R=-0.61,P <0.01). Estimators of species richness (Observed species, ACE and Chao1) were all strongly positively correlated with T (all R >0.52, P <0.05) but negative with DO (all R <-0.64, P <0.05) (Supplementary Table S2). In particular, depth and DO were significantly negatively associated with some of the bacterial genera, such as Tenacibaculum, Ponticaulis,Arcobacter, Oleispira, and Sinobacterium, while temperature was strongly positively correlated with them (Fig.8a). Another environmental factor salinity was significantly positively correlated Tenacibaculum,Arcobacter, and Thalassospira, but possibly significantly negatively aff ecting the diversity of Oceanicaulis. In addition, nitrite was significantly negatively correlated with Colwellia and Oleispira,while positively correlated with Pseudophaeobacter.
For Archaea, Spearman’s rank correlation revealed that the Shannon value was positively correlated with T ( R=0.74, P <0.01) and salinity ( R=0.83, P <0.01) but negatively with depth ( R=-0.82, P <0.01) and DO( R=-0.78, P <0.01). Moreover, species richnessindices were also significantly positively correlated with T (all R >0.48, P <0.05), while Observed species and Chao1 were negatively correlated with DO (all R <-0.65, P <0.05) (Supplementary Table S2). We also found that T and salinity were negatively correlated with unidentified Thermoplasmata; however, depth and DO had the strongest influence on it (Fig.8b).
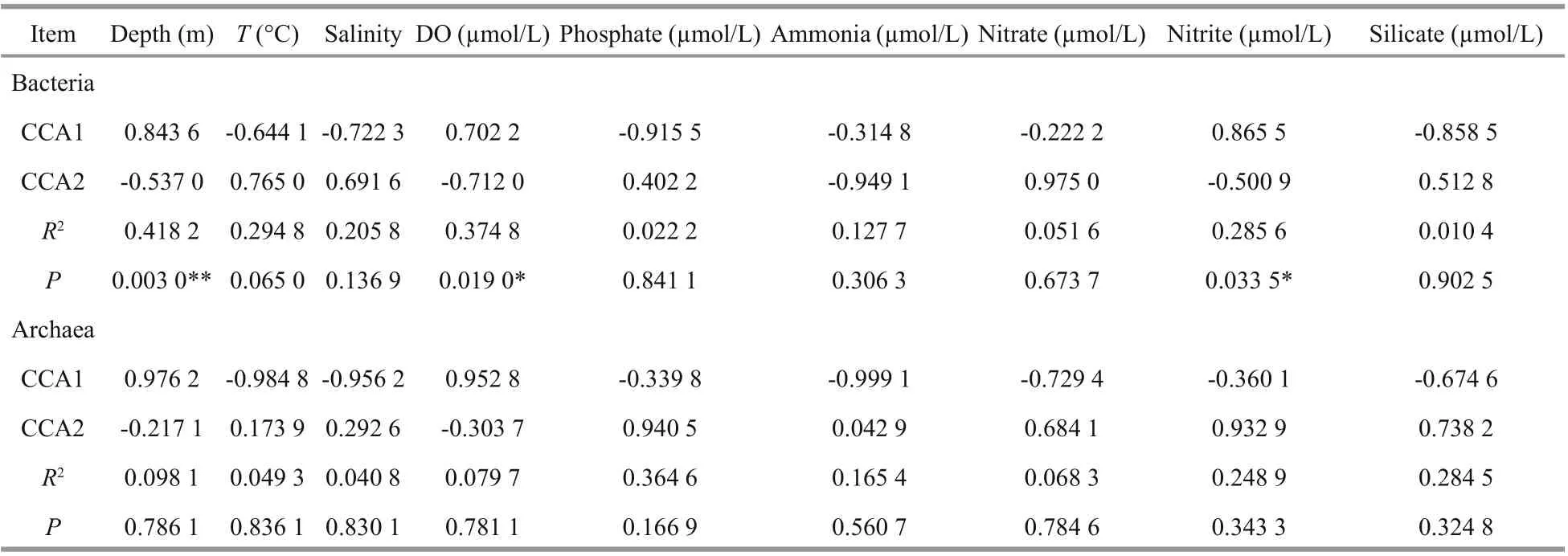
Table 3 CCA’s rank order correlation analysis
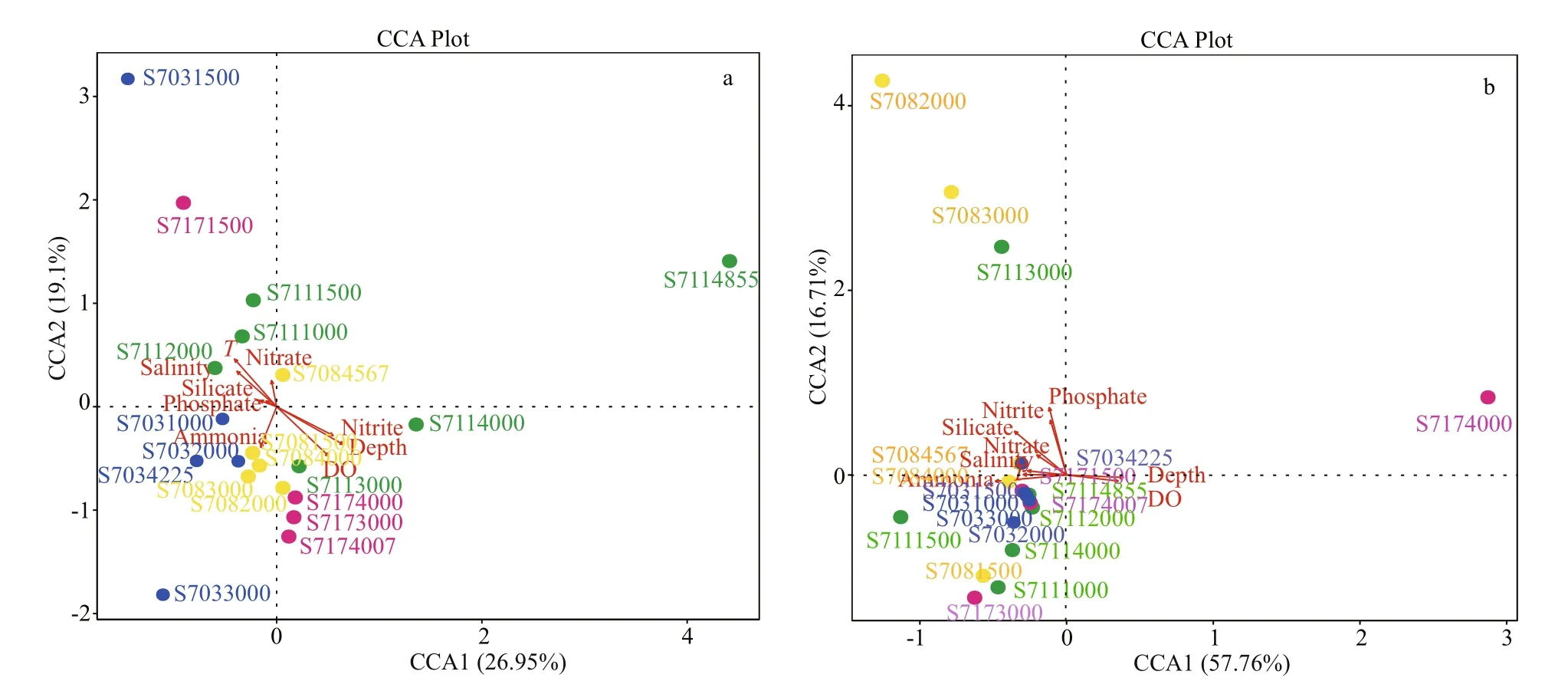
Fig.9 Canonical correspondence analysis (CCA) biplot of the distribution of bacterial (a) and archaeal (b) species at the genus level with environmental factors
Bacterial community dynamics with respect to the environmental variables were analyzed using CCA. It revealed that the first and second axes together explained 46.05% of the cumulative variance of the bacterial community at the genus level (Fig.9a), with the corresponding values of 0.003 0 for depth, 0.019 0 for DO, 0.033 5 for nitrite, 0.065 0 for temperature and >0.13 for other parameters (Table 3). All these indicated that diff erent environmental factors had varying eff ects on the bacterial taxa, and among the variables, it was depth, DO and nitrite that had the strongest influence on the structure of the bacterial communities.
Further, the first two CCA axes accounted for 74.4% of the explained total variance, it seems that the archaeal communities at genus level were heavily influenced by the above mentioned environmental variables but none of them exhibited significant relationships ( P >0.1) (Fig.9b, Table 3). However,depth, temperature, salinity, and DO were shown to be positive/negative factors aff ecting the distribution of several archaeal genera revealed by Spearman analysis.
4 DISCUSSION
The bacterial richness (mean Chao1 of 359.0) in the studied area was much lower than those previously reported in the subsurface seawater from coastal ecosystem of Arabian Sea (814.20) (Kumar et al.,2019) and the deep-sea sediments from Southern Ocean (606) (Jamieson et al., 2013). Similarly, ACE values we observed (mean ACE of 352.5) were also lower than those reported at 1 000 m in water samples from East China Sea (3073) (Dong et al., 2014),biofilms (1469) (Guo et al., 2017) and intertidal sediments (4236) (Guo et al., 2018) collected from the Changjiang River estuary. In addition, the bacterial diversity indices for the deep-sea water in this study(3.53) was lower than those reported in studies on the subsurface seawater from coastal ecosystems of Arabian Sea (3.77) (Kumar et al., 2019), seawater samples (4.72) and sediments (7.65) from the East China Sea (Dong et al., 2014), sediments (5.96) from the Changjiang River estuary (Feng et al., 2009).These results showed that the richness and diversity of bacteria from coastal, subsurface water and intertidal sediment environments were higher than that of the deep-sea waters in pelagic ocean, which could be due to higher nutrients from inland sources.Furthermore, previous study has reported the inhibition of upwelling and mixing of the deeper waters, making the Eastern Equatorial Indian Ocean oligotrophic (Kumar et al., 2009).
In general, Proteobacteria dominated in all 4 stations in this area, consistent with other studies in deep-sea waters collected from the northern Indian Ocean (Menezes et al., 2018), western Arctic Ocean(Han et al., 2015), North Atlantic Ocean (Sogin et al.,2006; Agogué et al., 2011) and North Pacific Ocean(Shulse et al., 2017). Within the Proteobacteria, the relative abundance of the major taxa (alpha-, beta-,gamma- and delta-Proteobacteria) were remarkably distinct in the diff erent sea waters from subsurface to bathypelagic zones. Compared to bacterial communities in deep seawater collected from western Arctic Ocean (Han et al., 2015) and North Atlantic Ocean (Sogin et al., 2006) with predominant class of Alphaproteobacteria, bacterial communities in the Ninetyeast Ridge were mostly dominated with Gammaproteobacteria (37.51%-88.55%), followed by Alphaproteobacteria (4.62%-45.03%) (Fig.6a),which is consistent with the report on the sea water(5-1 000 m) collected from Arabian Sea Oxygen Minimum Zone (OMZ) (Bandekar et al., 2018).Betaproteobacteria has been measured the most abundant taxon in the sediment (Guo et al., 2018),however, it was not present in this water column. It is reported that the Eastern Equatorial Indian Ocean was a typical oligotrophic area (Kumar et al., 2009),coincidently, Gammaproteobacteria have been reported to adopt to oligotrophic environments with wide temperature range tolerance (Cho and Giovannoni, 2004). Within Alphaproteobacteria,Donghicola and Sulfitobacter were the most abundant genera belonging to Rhodobacteraceae in the water mass. It is reported that Rhodobacteraceae cooccurred with nitrogen metabolism and stress response. Meanwhile, Rhodobacteraceae correlated with amino acids and derivatives, indicating potential for further delineation of oligotrophs based on metabolic requirements (Haggerty and Dinsdale,2017).
Bacteroidetes was the second most abundant phylum in all the samples examined, consisting of 1 class, 5 orders, and 20 families. Flavobacteriaceae was the only family detected in all of the 20 deep-sea samples and included one genus Marinobacter in the top 20 (Fig.7a). Previous report has shown that Flavobacteriaceae was essential in organic matter remineralization in the environment (Cottrell et al.,2005), because they can secrete hydrolytic enzymes and decompose various complex biopolymers(Williams et al., 2013). On the other hand,Actinobacteria, which is considered significant contributors to carbon storage in terrestrial ecosystems(Freedman and Zak, 2014), was found ubiquitous in the deep-sea environments in this study. Thus, these suggest that these bacteria may have been enriched because of the deposition of the organic matter in the deeper or bottom waters. In addition, Cyanobacteria are oxygenic photosynthetic prokaryotes, which prefer the sunlit surface waters than the aphotic waters below 1 000 m. However, it was surprising to observe that the relative abundance of Cyanobacteria (18.93%)in the 4 000 m of station S711 was significantly higher than the other samples. Thus, further research focused on this phylum is needed.
In contrast to bacteria, less is known about archaeal community structure in the deep-sea environments of the Ninetyeast Ridge. Euryarchaeota, Crenarchaeota,and Thaumarchaeota are known to be the three major archaeal phyla in many environments globally. In this study, only Thaumarchaeota and Euryarchaeota were identified in the deep-water column. Specifically,Thaumarchaeota was the most abundant phylum with the relative abundance range from 67.3% to 97.9% in the 20 seawater samples. Previous studies have shown that Thaumarchaeota established in the year of 2008(formerly Marine Group I, Crenarchaeota) (Brochier-Armanet et al., 2008) were globally abundant aerobic chemolithoautotrophs. They generate energy by oxidizing ammonia to nitrite (Könneke et al., 2005)and have been recognized as the dominant nitrifiers in a wide range of terrestrial and marine habitats.Physiological, genetic and biochemical studies on isolated Thaumarchaeota suggest that marine planktonic Thaumarchaeota are well adapted to oligotrophic conditions due to their high substrateaffi nities (Martens-Habbena et al., 2009) and an energy effi cient carbon fixation pathway (Könneke et al., 2012), typically encountered in the pelagic oceanic habitats. Thaumarchaeota are known aerobic ammonium oxidizing Archaea (Stahl and de la Torre,2012; Off re et al., 2013), and their ubiquity in the study area indicates a possibly frequently occurring ammonia-oxidizing process and carbon cycling in the deep-sea water around the Ninetyeast Ridge. Within Euryarchaeota, sequences clustering among unclassified order within the class Thermoplasmata demonstrated the greatest relative abundance in this area, with relative abundances decreasing with depth in the water column. Thermoplasmata were reported played an important role in anaerobic environment with limited electron acceptors such as oxygen and sulfate. Additionally, Thermoplasmata collected from the stomach of cattle involved in the methane consumption process at medium temperature in the previous study. It can be concluded that Thermoplasmata were probably related to methane metabolism in deep seawater (Lin et al., 2015; Xie et al., 2017).
Environmental conditions in the ocean are variable associated with geographical isolations and environmental factors, which could then be responsible for the observed microbial community patterns. Previous study reported that latitude was the most significant factor explaining the variations in bacterial communities at higher taxonomic levels in the north Chinese marginal seas (Liu et al., 2015). In this study, the bacterial communities at S703 and S708 clustered together, while S711 and S717 also grouped into another cluster except for 3 000 m of S717, confirming that geographical distance may be an important factor aff ecting bacterial communities in the area (Figs.4 & 5).
UPGMA clustering also showed that the archaeal communities were divided into three clusters along the vertical gradient. The samples from depths<2 000 m formed one cluster, while samples from 3 000 m except for station S717 also formed another group, and >4 000 m forming the third cluster. These suggest that water depth may be responsible for the vertical similarity of the microbial communities at the study sites, as revealed by NMDS analysis.Meanwhile, depth was confirmed to be negatively associated with some of the bacterial and archaeal genera, because the growth and activity of these microbes were closely related to water temperature and pressure, which greatly changed with depth.
Several studies have found that microbial community composition and structure were often aff ected by nutrients including phosphate, ammonia,nitrate, and nitrite (Biddanda et al., 2001). For instance, nitrate concentration has been reported to be positively correlated with bacterial anammox rate(Brin et al., 2014). The eastern Indian Ocean is a typical oligotrophic environment, some anammox bacteria had been noted to be suitable to oligotrophic environments (Rysgaard et al., 2004). In this study,results of Spearman’s correlation and CCA analysis at genus level revealed that nitrite was one of the significant environmental factors ( P=0.033) that could explain variations in the bacterial community structures. And among the bacterial genera detected in this present study, nitrite was specifically significantly correlated with several genera, such as Colwellia, Oleispira, and Pseudophaeobacter(Fig.8a). This confirms that nitrite may play an important role in structuring bacterial communities in the deep sea.
Finally, DO is another potential factor regulating the distribution of bacteria. In this study, DO increased with depth in deep sea water, which was consistent with the previous reports in Indian Ocean (Naqvi,2006; Paulmier and Ruiz-Pino, 2009). In the deep cold waters, the low temperature increased the solubility of oxygen in seawater. Moreover, due to the decreased flux of export production in deep waters,the oxygen demand associated with the remineralization of sinking organic matter was relatively weak. As a result, the oxygen content usually presented an increasing trend as the water depth increased in deep waters (Paulmier and Ruiz-Pino, 2009). In addition, Oxygen-deficient zones(ODZs) had been demonstrated to serve as unique habitats for microbial communities (Beman and Carolan, 2013). Similarly, oxygen minimum zones(OMZs) also provide unique habitats. Compared to the surface communities, which exhibit seasonal variability, communities in the OMZ tend to be less variable (Bandekar et al., 2018). The concentrations of DO seem to result in the vertical separation among bacterial communities, consistent with our results.More focused studies on the responses of microbial communities to DO at diff erent depths-from surface to bathyal zone-in the Ninetyeast Ridge are needed.
5 CONCLUSION
Results of this study demonstrate that both bacterial and archaeal communities were diverse, and dominant groups varied among the stations and depths.Gammaproteobacteria and Alphaproteobacteria dominated the bacterial communities, while Nitrososphaeria and Thermoplasmata for the Archaea.Since the eastern Indian Ocean is a typical oligotrophic environment, the richness and diversity of bacteria and Archaea were lower than those reported in other environments. Depth, DO and nitrite could have significantly aff ected the structuring of bacterial communities, whereas archaeal communities were also heavily influenced by above-mentioned environmental variables but none of them in this study displayed any significant correlation ( P >0.1). Lastly,our study suggests that ammonia-oxidation and carbon cycling may be occurring more than expected in the deep-sea water around the Ninetyeast Ridge.
6 DATA AVAILABILITY STATEMENT
The original data generated or analyzed during the current study are available from the corresponding author on reasonable request.
杂志排行

Journal of Oceanology and Limnology的其它文章
- Predicting sediment flux from continental shelfislands,southeastern China*
- Laboratory simulation of dissolved oxygen reduction and ammonia nitrogen generation in the decay stage of harmful algae bloom*
- Development of high-resolution chloroplast markers for intraspecific phylogeographic studies of Phaeocystis globosa*
- Effects ofiron and humic acid on competition between Microcystis aeruginosa and Scenedesmus obliquus revealed by HPLC analysis of pigments*
- Effect of river plume on phytoplankton community structure in Zhujiang River estuary*
- Exploring the sublethal genotoxic effects of class II organophosphorus insecticide quinalphos on freshwater fish Cyprinus carpio