太湖流域地方猪种遗传结构评估和体重体尺性状差异相关选择位点鉴定
2021-04-13刘晨曦侯黎明周五朵徐燕尹彦镇周天威李平华黄瑞华
刘晨曦,侯黎明,周五朵,徐燕,尹彦镇,周天威,李平华*,黄瑞华*
(1.南京农业大学养猪研究所,江苏 南京 210095;2.南京农业大学淮安研究院,江苏 淮安 223005;3.江苏省畜牧总站,江苏 南京 210036)
中国太湖流域地方猪种包括二花脸猪(EHL),梅山猪(MS),枫泾猪(FJ),米猪(MI),嘉兴黑猪(JXB),沙乌头猪(SWT)和红灯笼猪(HDL)。其中,梅山猪根据体型大小分为中型梅山猪(MMS)和小型梅山猪(SMS)两个亚群[1]。这些猪种均具有耐粗饲,优秀的肉质和出色的繁殖性能[2-4],为商品猪的持续选育提供了宝贵的遗传资源[2]。
太湖流域地方猪独特的表型,意味其可能具备独特的遗传结构。Chen等[5]研究发现,太湖流域地方猪各品种及梅山猪品种内两个亚群在聚类模式上可以相互区分,说明各个群体间的遗传起源上独立,结构并不完全一致[5-7]。并且发现米猪和二花脸猪之间遗传关系更为接近[6]。
研究人员利用太湖流域地方猪种鉴定到多个与体重体尺相关的候选基因(QTLs),SUN等[8]在梅山猪两个亚群中鉴定到SPDEF,PACSIN1等与体重相关的基因在这两个亚群间受到不同方向的选择,用皮特兰和梅山猪杂交的F2代群体,鉴定到3号染色体影响日增重的QTL[9],以及影响该F2代群体的生长性状和胴体性状的FTO基因[10]。已有研究对象没有涵盖该地区地方猪种现存的全部血统,并且关于太湖流域地方猪各品种及梅山猪品种内两个亚群间体重体尺差异的遗传机制的研究不充分,没有利用其他遗传结构接近但体型差异显著的猪种进行验证分析。为了解决以上问题,本研究采集了440头代表太湖流域地方猪最全面血统的猪群样本,利用猪50 K芯片获得每个样本的基因分型数据,对基因分型数据集进行群体结构分析和选择位点鉴定。本研究结果全面解析了太湖流域地方猪群体结构,并可为深入解析这些群体体重体尺性状差异提供了理论基础。
1 材料与方法
1.1 试验动物
采集440头二花脸猪(EHL),枫泾猪(FJ),米猪(MI),嘉兴黑猪(JXB),沙乌头猪(SWT),红灯笼猪(HDL),中型梅山猪(MMS)和小型梅山猪(SMS)的耳组织样。二花脸猪(n=111)分别从常州市,常熟市和苏州市的保种场采集,小型梅山猪(n=71)分别从太仓市和苏州市的保种场采集,中型梅山猪(n=65)从昆山市保种场采集,枫泾猪(n=23)从苏州市的保种场采集,米猪(n=59)从常州市的保种场采集,嘉兴黑猪(n=40)从嘉兴市的保种场采集,沙乌头猪(n=41)从南通市的保种场采集,红灯笼猪(n=30)从溧阳市的保种场采集。这440头个体可以涵盖江苏省太湖流域地方猪保种群的全部血统。采样的所有公猪和母猪在3代内没有共同血亲。
1.2 个体基因组信息鉴定和收集
将440头猪耳组织样本用常规的苯酚/氯仿方法提取DNA,并稀释至最终浓度50 ng/μL,然后采用北京康普森公司研发的猪50 K基因型芯片(测序平台为美国Illumina公司提供)进行基因分型,芯片总计含有51 315个SNP位点。基因型输入文件转换为PLINK v1.07格式文件[11]用于后续分析。
对PLINK格式文件进行SNP位点质控,具体条件为:单个个体SNP位点检出率大于90%,每个SNP检出率大于90%,微小等位基因频率大于5%。所有未知位置信息和性染色体上的SNP位点都剔除。经过质控,获得总计29 754个满足条件的SNP位点。此外,采用上述SNP位点质控原则,在65头中型梅山猪和71头小型梅山猪群体内部进行质控,经过质控得到总计21 850个有效SNP位点;在111头二花脸和59头米猪个体进行质控并得到29 754个有效SNP位点。
1.3 群体间遗传距离(1-Dst)计算
为了估计个体间的遗传距离,使用PLINK v1.07软件计算了等位基因共享比例(Dst)[11]。所有个体之间的遗传距离计算公式为(1-Dst)[12]。基于(1-Dst)矩阵,使用MEGA v6.0软件[13]计算两两群体之间平均遗传距离的大小。使用群体间遗传距离(1-Dst)矩阵构建聚类树热图,通过内部R脚本进行可视化。
1.4 群体间遗传分化系数(Fst)计算
使用Wright’s F-derived统计法来计算品种间遗传分化系数Fst[14],使用GENEPOP v4.2软件[15]进行相关计算。不同的Fst值代表的意义为:Fst在0~0.05表示较低的遗传分化,Fst在0.05~0.15表示中等的遗传分化,Fst在0.15~0.25表示较高的遗传分化,Fst高于0.25表示高度遗传分化[16-17]。使用群体间遗传分化系数(Fst)矩阵构建了聚类树热图,并通过内部R脚本进行可视化。
1.5 群体结构分析
基于SNP基因型数据,使用ADMIXTURE v1.23软件[18]推断出最可能的祖先血统组成(K),即假定有K个祖先存在的前提下,每个群体内含有的各祖先血统的比例,根据最小交叉验证误差(CV-error)的原则确定最大K值。对于太湖流域地方猪,使用内部R脚本将K=2~8的结果进行可视化。为了增加ADMIXTURE分析的准确性,并避免由这些群体的不同样本量引起的潜在偏差,我们从每个群体中随机选择了23头个体进行ADMIXTURE分析。
1.6 受选择SNP位点鉴定
使用GENEPOP v4.2软件[16]进行中型梅山猪和小型梅山猪群体间Fst分析,每个SNP位点计算得到相应遗传分化(Fst)值。对于所有SNP位点的Fst值,采用Z检验,鉴定极显著(P<0.01)偏离整体Fst均值的SNP位点[19],并将这些SNP位点作为在梅山猪亚群间受选择的候选SNP位点。采用相同计算原理鉴定二花脸和米猪群体间受选择的候选SNP位点。最终,将梅山猪亚群间候选SNP位点与二花脸和米猪群体间候选SNP位点取交集,作为更有可能的受选择位点。
1.7 候选基因注释
对SNP位点上下游各50 kb[20]的区域内进行基因注释,使用猪11.1版本参考基因组(Sscrofa 11.1),注释网站为ensemble官网(http://www.ensembl.org)。
1.8 受选择SNP位点间连锁不平衡分析
在人为或自然选择的压力下,优势等位基因型频率会不断上升,并且由于“搭车效应”形成选择性清除区域,选择性清除区域附近的SNP位点连锁不平衡程度加深[21]。为了进一步确定候选SNP位点受选择的可能性,我们使用PLINK v1.07软件[13]来检测位于同一染色体且位置距离接近的多个受选择SNP位点间的连锁不平衡情况(r2)。执行命令“Plink-file-r2”,通过SNP位点间的相关系数r2值来估算连锁不平衡(LD)程度,并以r2值0.3作为阈值来衡量位点间是否存在连锁不平衡[12,22]。
1.9 受选择SNP位点与猪体重体尺等性状QTLs注释
对于1、3和6号染色体受到选择的SNP位点形成的受选择区域(相应染色体彼此位置距离接近且强连锁的多个受选择SNP位点,以位置距离最小的SNP位点作为起点,位置距离最大的SNP位点作为终点,形成的受选择区域)进行猪体重体尺等相关性状QTLs注释,注释网站为PigQTLdb官网(https://www.animalgenome.org/cgi-bin/QTLdb/SS/index)。
2 结果与分析
2.1 太湖流域地方猪群体内部结构
2.1.1 太湖流域地方猪各品种及梅山猪品种内两个亚群间遗传关系
由表1可知,在太湖流域地方猪各品种及梅山猪品种内两个亚群间的遗传距离(1-Dst)分析中,群体间遗传距离最为接近的是二花脸和米猪,群体间遗传距离为0.100,其次较为接近的是中型梅山猪与小型梅山猪,群体间遗传距离为0.153。二花脸和米猪间的遗传距离比梅山猪两个亚群更为接近。

表1 太湖流域8个地方猪群体间遗传距离(1-Dst)
8个地方猪在群体间遗传分化方面的分析结果如表2所示。中型梅山猪与小型梅山猪群体间的遗传分化程度最小,遗传分化系数为0.264,其次为枫泾和嘉兴黑间的遗传分化系数,数值为0.278,排在第三的是二花脸和米猪间的遗传分化系数,数值为0.282。所有群体间的遗传分化系数都超过0.250。

表2 太湖流域8个地方猪群体间遗传分化系数(Fst)
2.1.2 太湖流域地方猪各品种及梅山猪品种内两个亚群间ADMIXTURE分析
为了判定太湖流域地方猪各品种及梅山猪品种内两个亚群间的进化路线,进行了ADMIXTURE分析,结果如图1所示。图1展示了K值为2~8的结果,在K值为8时,ADMIXTURE分析拥有最小的交叉检验误差(CV-error),梅山猪两个亚群的K值为2~5,这两个群体都保持着一致的祖先血统组成,K值为6时,分化成两个不同的群体。二花脸和米猪的K值为2~7时,这两个群体都保持着一致的祖先血统组成,直到K为8时才最终分化成两个群体。二花脸和米猪拥有最为一致的进化路线,这与二花脸和米猪间遗传距离最小的结果一致。此外,枫泾和嘉兴黑间的祖先血统组成在K值为2~5时保持一致。在K值为8(最小CV-error值)时,这8个群体都可以区分成不同的群体。

注:EHL代表二花脸猪;MMS代表中型梅山猪;SMS代表小型梅山猪;FJ代表枫泾猪;HDL代表红灯笼猪;JXB代表嘉兴黑猪;MI代表米猪;SWT代表沙乌头猪;每种颜色代表一个祖先群,群体间均以虚线分隔图1 太湖流域地方猪8个群体的祖先血统组成
2.2 太湖流域地方猪受选择SNP位点鉴定
2.2.1 梅山猪亚群间以及二花脸猪和米猪群体间共同分化SNP位点鉴定及基因注释结果
对梅山猪亚群间、以及二花脸和米猪群体间进行两组Fst分析,鉴定这两组共同分化的SNP位点,并作为太湖流域地方猪受选择的候选SNP位点。将阈值线以上的点作为群体间显著分化的SNP位点(Z检验,P<0.01),结果如图2所示。一共发现24个SNP位点在这两组Fst分析中都显著分化,其中,有7个位于1号染色体,6个位于3号染色体,6个位于6号染色体,彼此间距离接近,其余分布在2、4、5和16号染色体。
对这24个候选SNP位点进行基因注释,得到20个可以编码蛋白的基因,其中有8个基因与体重体尺性状相关,具体信息见表3。这些基因主要有如下功能:体重体尺(DENND1A、LHX2、NR6A1、FANCL、KCNK3、CDH11、CDH5和AGBL4),繁殖性状(STRBP、FANCL、MRGPRF和PSME4),神经发育(MVB12B、ARHGAP39、CNTN1、PLCG2和CTNND2),抗病力(NEK6和SCAI),嗅觉(NR2C1),运动能力(VPS13D)。
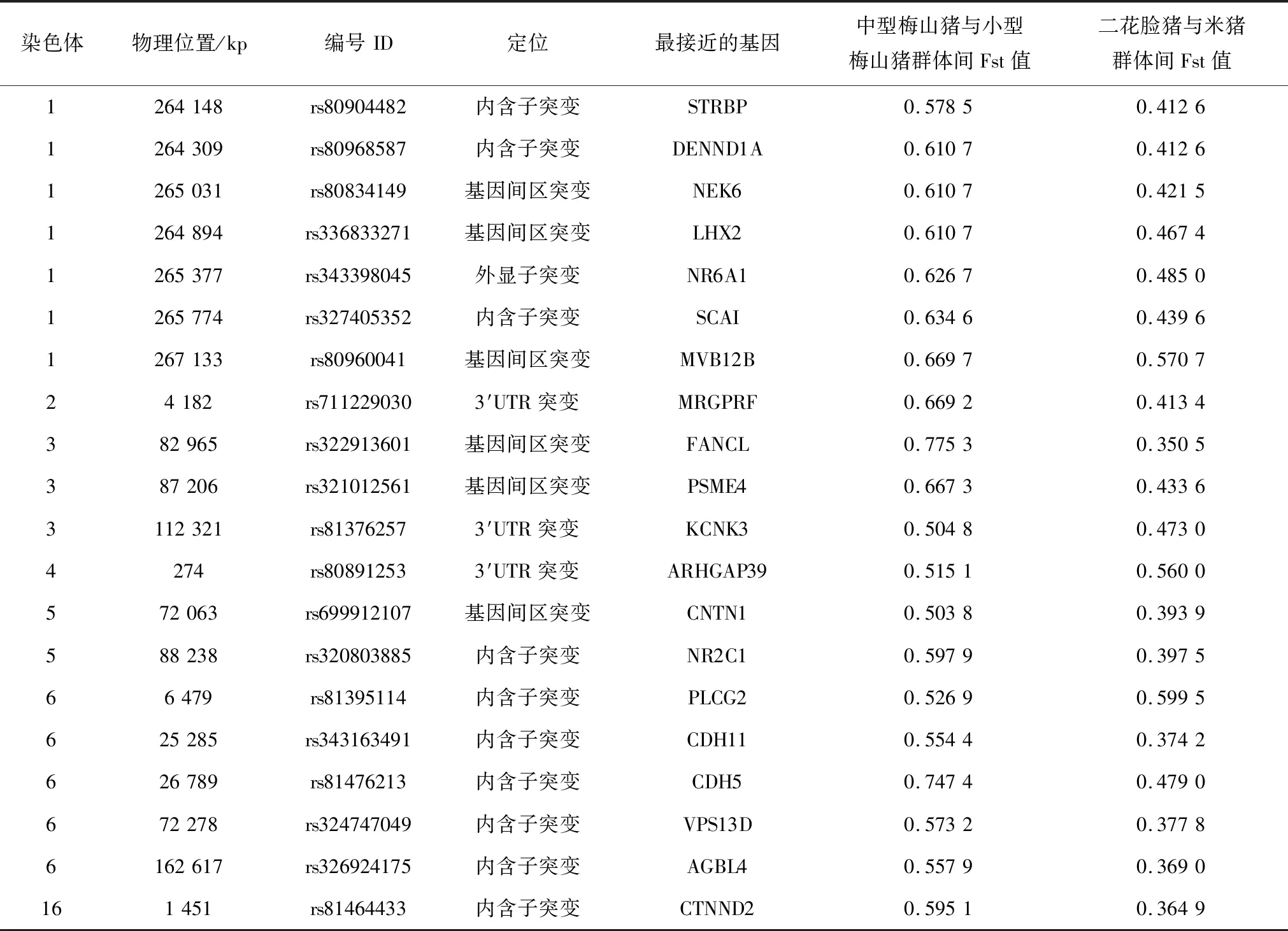
表3 受选择SNP位点基因注释结果
2.2.2 受选择SNP位点间连锁不平衡分析
针对1、3和6号染色体中受选择候选SNP位点,多数位置距离彼此接近,因此我们对这3条染色体上的位置距离彼此接近的候选SNP位点进行连锁不平衡分析,结果见表4。上述3条染色体内的候选SNP位点都存在较强的连锁不平衡,r2值远大于0.3,其中1号染色体多个SNP位点达到完全连锁,r2值为1。并且1号染色体彼此位置距离接近的7个SNP位点,分别在中型梅山猪和二花脸群体中形成了一种主要的单倍型“TCCGAAA”和“TCCGAAA”,同时在小型梅山猪和米猪群体内变异较低;对于3号染色体彼此位置距离接近的5个SNP位点,分别在中型梅山猪,小型梅山猪,二花脸和米猪群体中形成了一种主要的单倍型“GGCAG”,“AATGA”,“GGCAG”和“AATGA”;对于6号染色体彼此位置距离接近的3个SNP位点,分别在中型梅山猪和米猪群体中形成了一种主要的单倍型“GTG”和“ACA”。可见1、3和6号染色体在群体间经历了不同方向的选择,导致群体间分化增大,并使得群体内变异程度减少,逐渐形成单一的单倍型。
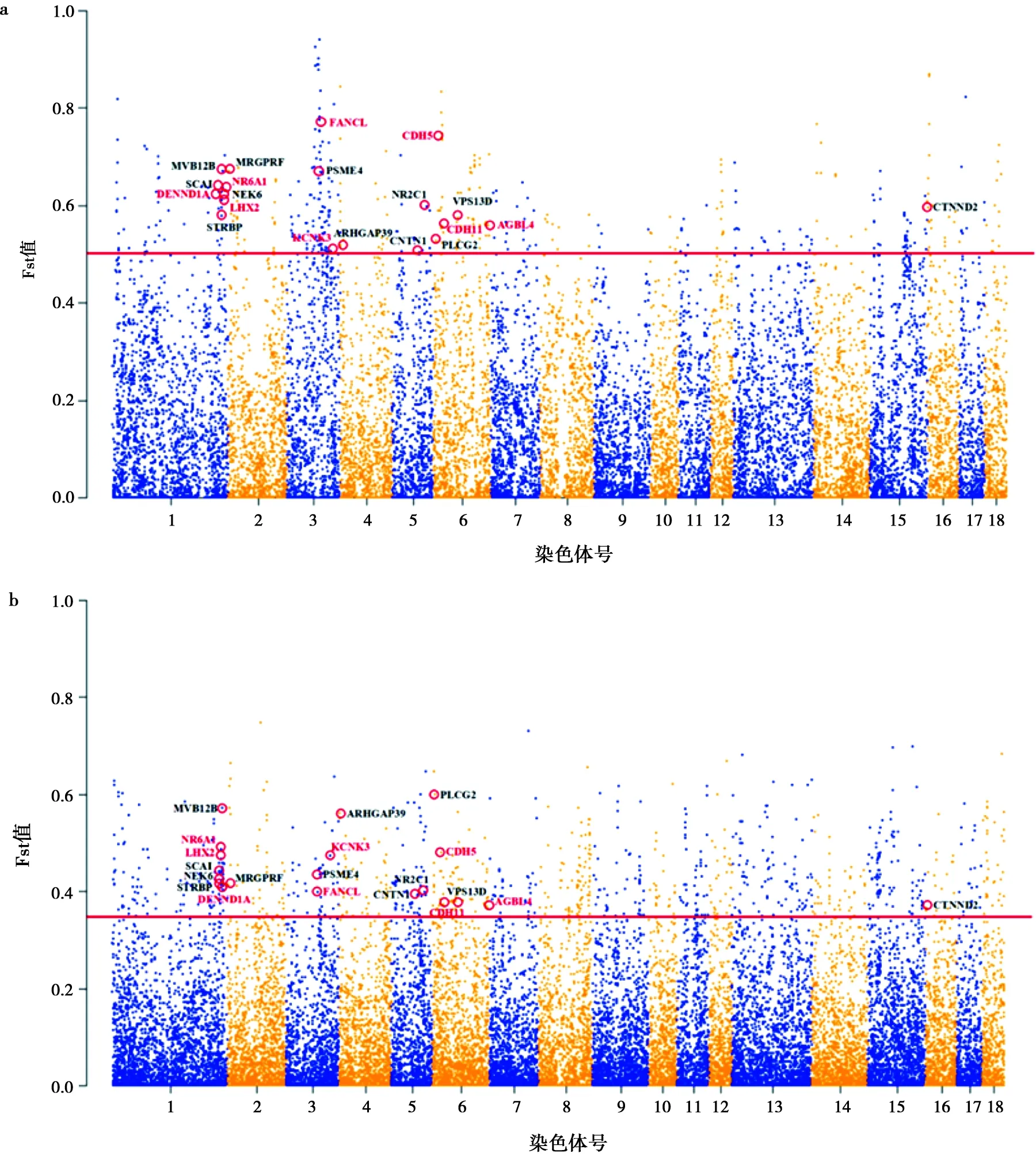
a.中型梅山和小型梅山群体间Fst分析;b.米猪和二花脸群体间Fst分析;x轴为18条染色体,y轴为每个位点Fst值;不同的染色体用不同颜色标注,红色实线为阈值线,阈值(Z检验,P<0.01)上的点代表受选择位点,候选基因用红色圆框标注,与体重体尺相关的候选基因用红色字体标注图2 梅山猪亚群间以及二花脸猪和米猪群体间共同分化SNP位点鉴定
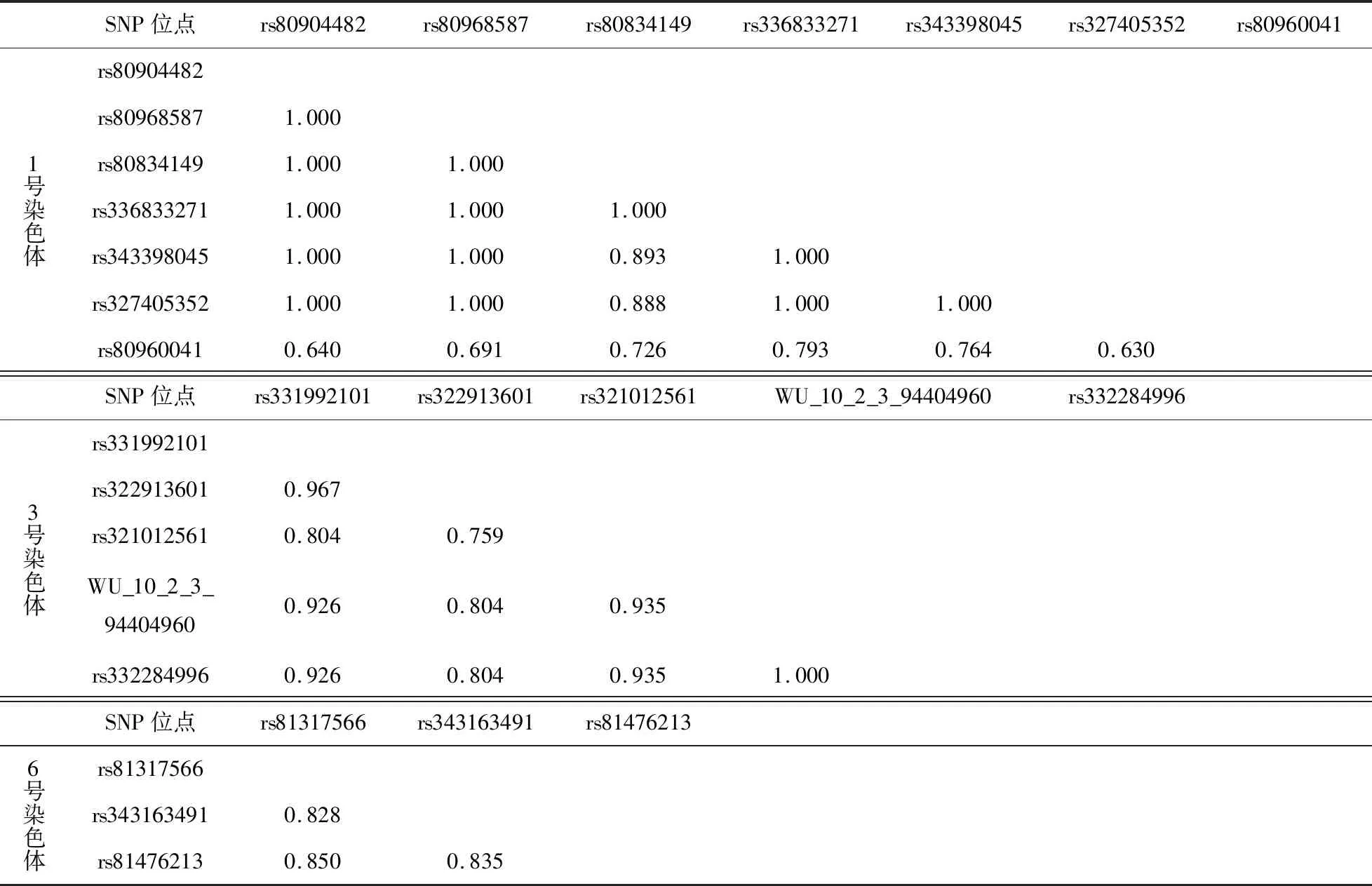
表4 猪1号、3号和6号染色体受选择SNP位点间的连锁不平衡分析(r2)
2.2.3 受选择SNP位点与猪体重体尺性状相关QTLs注释
对SNP位点所形成的选择区域进行猪体重体尺等相关性状QTLs注释,结果见表5。1、3和6号染色体相应区域注释到大量的QTLs与猪的体重体尺性状相关,其中主要包括猪日增重性状,体重性状,椎骨数性状,料重比性状和采食量性状,表明这些区域很可能对于猪的体重体尺等表型的形成起到巨大作用。
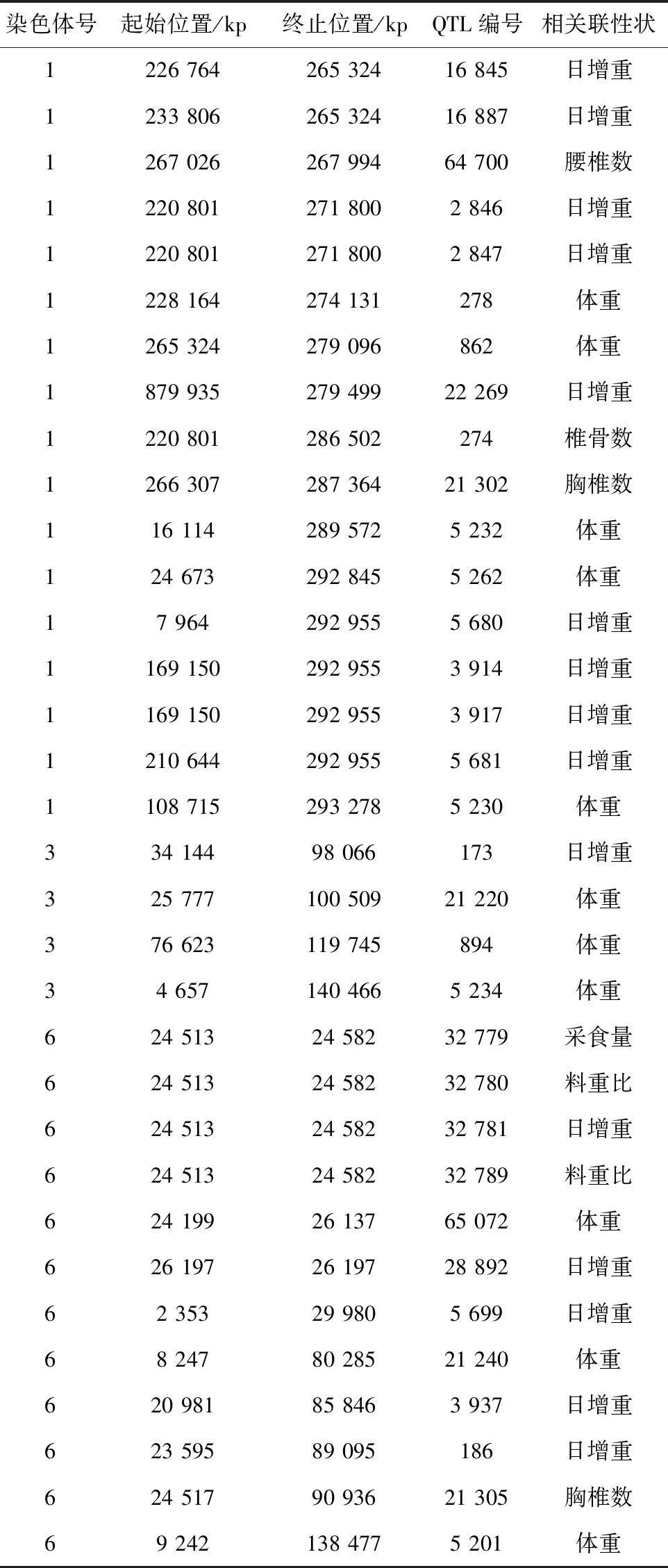
表5 受选择SNP位点所在区域QTLs注释结果
3 讨论
3.1 太湖流域地方猪种内部结构研究
通过对太湖流域地方猪各品种,以及梅山猪品种内两个亚群进行多种群体遗传学分析发现,梅山猪亚群间遗传距离接近,遗传分化程度最低,进化路线相对一致,而二花脸与米猪间也有着最近的遗传距离,较低的分化程度以及最一致的进化路线,这与他人的研究结果一致[6,24]。试验发现,二花脸猪与米猪之间的遗传距离小于梅山猪两个亚群间的遗传距离,并且拥有最为一致的祖先血统组成,表明二花脸猪与米猪之间的遗传关系最为接近,其接近程度甚至超过了梅山猪的两个亚群间的遗传关系,该结果与Wang等[6]研究一致。此外还发现,所有太湖流域地方猪群体间的遗传分化都超过0.25,达到高度分化水平[18-19],并在ADMIXTURE分析中,K值为8时(最小的CV-error),各自形成了不同的祖先血统组成情况,可见包括梅山猪两个亚群在内的8个群体彼此间都存在一定的分化,因此在保种时应分开保种。
3.2 太湖流域地方猪种体重体尺性状差异的候选SNP位点鉴定
对于梅山猪亚群、二花脸和米猪这两个群体组合分别进行遗传分化Fst的分析,并寻找在两组分析中共同分化的SNP位点作为候选SNP位点。采用这种遗传结构十分接近、但体重体尺性状差异显著的群体进行比对分析,可以减少由于群体间其他性状差异或者本身遗传结构差异导致SNP位点显著分化的可能性。为了减少显著分化SNP位点是假阳性的概率,通过取两个分析结果交集的方法来进一步缩小差异SNP位点的范围。本试验共发现24个候选SNP位点,它们在这两组分析中都显著分化,这些候选SNP位点分布集中,主要分布在1、3、6号染色体区域内,表明1、3、6号染色体在这4个群体中更可能受到强的选择。
将受选择SNP位点进行基因注释,注释到20个可以编码蛋白的基因,8个基因被报道与体重体尺等性状相关。DENND1A基因的rs10818854位点与人的肥胖显著相关[25];LHX2基因通过调节破骨细胞中的RANKL信号传导调节小鼠的骨重塑[26];NR6A1基因作为猪肋骨数的主效基因,被运用于多个猪种的分子标记辅助选育改良过程中[27-28];FANCL基因可能是导致人肥胖的候选基因[29];KCNK3基因可以调节脂肪生热和肥胖[30];CDH11基因是调节小鼠骨骼结构和股骨形态的主要候选基因[31];髋臼唇细胞中的CDH5可能参与了骨关节炎的发病机制[32];AGBL4基因被发现与牛的生长等数量性状显著相关[33]。
对上述3个染色体的候选SNP位点进行连锁不平衡分析结果发现,每条染色体上候选SNP间的r2值都远大于0.3,达到强连锁水平[12,22],并且这些SNP在各个群体中都展现出较低多态性,形成了主要的单倍型。在人为或自然选择的压力下,优势等位基因型频率会不断上升,并且由于连锁不平衡,使得优势基因型附近形成大片段的纯合单倍型[19],而1、3、6号染色体内候选SNP位点间有很强的连锁不平衡,并且候选SNP位点多态性降低,表明这些候选SNP位点所在的区域受到较强的选择。
将受选择SNP位点与猪QTLs数据库进行比对,结果注释到大量与体重体尺性状相关的QTLs。Christine等[9]利用梅山猪和皮特兰杂交产生的F2代群体鉴定到1号染色体226 764~265 324 kp处存在影响日增重的QTL;HUANG等[33]利用杜洛克和二花脸杂交产生的F2代群体鉴定到1号染色体266 307~287 364 kp处存在影响胸椎数的QTL;Guo等[34]利用大白猪和梅山猪杂交产生的F2代群体鉴定到3号染色体4 657~140 466 kp处存在一个大的QTL影响猪体重性状;利用皮特兰和梅山猪杂交产生的F2代群体鉴定到6号染色体24 513~24 582 kp处存在一个影响料重比的QTL[10]。这些结果都与本试验发现的候选SNP所在区域重合。以上证据表明,在两个Fst分析中共同受选择,且基因注释结果与体重和体尺性状相关的区域,可能与太湖流域地方猪种群体间体重体尺性状差异有关,这些区域将作为候选区域进行进一步研究。
致谢:常州市焦溪二花脸猪专业合作社及其工作人员顾岳清、黄媛和李顺等,常熟市牧工商有限公司、国家级二花脸猪保种中心及其工作人员陆志强等,苏州苏太企业有限公司及其工作人员黄雪根等,浙江青莲食品股份有限公司及其工作人员赵善林等,江苏省溧阳市乾丰养殖有限公司及其工作人员程跃强等,昆山市梅山猪保种有限公司及其工作人员沈永杰、时建新、周腾冰等,太仓市种猪场及其工作人员石晓峰、施擎天等,江苏兴旺农牧科技发展有限公司、国家级沙乌头猪保种场及其工作人员唐慧娟等,常州市金坛米猪原种场及其工作人员田国兴等,对于猪样本采集工作予以支持和帮助,在此一并感谢。
