程序性细胞坏死参与急慢性损伤脑内神经元死亡的研究进展
2021-04-09钱奕茗综述孙凤艳审校
钱奕茗(综述) 孙凤艳(审校)
(复旦大学基础医学院神经生物学系 上海200032)
细胞的死亡和新生是机体维持稳态环境的根本。1973年,Schweichel和Merker两位学者按照细胞死亡的形态学特点,将细胞死亡分为3大类:坏死(necrosis)、凋 亡(apoptosis)和 自 噬(autophagy)[1]。随着研究的逐步深入,新的细胞死亡方式和特征被不断发现。为此,2005年国际上成立了细胞死亡命名委员会(the Nomenclature Committee on Cell Death,NCCD)。NCCD专家根据细胞死亡方式不同提出细胞意外死亡(accidental cell death,ACD)和调节细胞死亡(regulated cell death,RCD)的分类法,并给出了新的定义。ACD是一类由各种伤害性刺激引起,且不受细胞内主动的程序性因素控制的细胞死亡方式,例如坏死;RCD通常为程序性细胞死亡过程,该类细胞死亡受到复杂的信号级联反应分子的调控。凋亡和自噬就属于RCD类型。近年来,以RCD形式的细胞死亡新类型被不断地发现,本文将介绍一种细胞坏死新的形式,即程序性细胞坏死。
传统认为,细胞坏死是一种被动的非程序性细胞死亡形式,常常伴有炎症的发生;而细胞凋亡和自噬则是一种主动的程序性细胞死亡过程[2]。随着细胞死亡相关研究的不断深入,人们发现有一类细胞死亡的形态特征与坏死相似,但是死亡过程类似凋亡的发生,即受到细胞内在信号分子的调节,抑制信号通路的药物能调节这类细胞死亡的发生发展[3]。由此改变了人们长期以来对坏死的定义,并命名这类受胞内信号分子调控的细胞坏死为necroptosis,即 取 坏 死necrosis字 首necro-,以 及apoptosis的字尾-ptosis而得名。本文意译暂称程序性细胞坏死。细胞膜上的死亡受体(Fas/TNF receptor associated factor/tumor necrosis factor receptor,Fas/TRAF2/TNFR等)激活可诱导程序性坏死[4],通过激活受体相互作用蛋白(receptor interaction protein,RIPK1/RIPK3)激酶及关键底物——混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein,MLKL)信号传导通路,使细胞膜完整性丢失、细胞器肿胀、线粒体功能障碍以及产生氧自由基等,导致细胞死亡[5-7]。深入研究表明,炎性细胞因子的分泌释放能激活程序性细胞坏死。开展对于程序性细胞坏死的研究和总结,能够帮助我们更好地理解相关疾病的病理过程,有助于疾病治疗的潜在靶点的研发。
程序性细胞坏死及其调节分子程序性细胞坏死的发生有两个重要的前提:(1)细胞能够表达RIPK3;(2)caspase-8活性受到抑制。启动程序性细胞坏死的受体有TNFR、Toll样受体3/4(Toll like receptors 3/4,TLR3/4)以及干扰素受体(interferon receptors,INFR)。病毒感染和药物等外界因素激活死亡受体可诱导其发生[8]。目前,人们对TNF-α介导的程序性细胞坏死研究最为详细。如图1所示,TNF-α与 受 体TNFR1相 结 合,在 胞 内 募 集RIPK1、TRAF2、细 胞凋亡抑 制蛋白(inhibitor of apoptosis proteins,cIAPs)等信号分子聚集,并在胞膜上形成复合物Ⅰ[9-10]。复合物Ⅰ依据接收的信号刺激对RIPK1进行不同修饰,从而决定细胞的存亡[11]。此时,caspase-8的作用至关重要。当caspase-8被激活时,RIPK3的活化被抑制,形成复合物Ⅱa,诱导细胞发生凋亡;然而,当caspase-8失活或缺失时,会促进RIPK1去泛素化,并与RIPK3、Fas相关死亡结构域蛋白(Fas-associating death domain-containing protein,FADD)结合,共同形成复合物Ⅱb。活化的RIPK1和RIPK3募集和磷酸化MLKL,激活下游信号转导通路,从而执行程序性细胞坏死[12-13]。目前认为,MLKL激酶的活化是执行程序性细胞坏死的重要环节。
MLKL是RIPK3激酶的底物,本身以无活性形式存在。MLKL的C末端区域含激酶结构域,N末端 含4螺 旋 结 构 域(four-helical bundledomain,4-HBD)。MLKL激酶的活化依赖于RIPK3诱导激酶结构域的残基磷酸化。人类MLKL的Thr357和Ser358及 小 鼠MLKL的Ser345、Ser347和Thr349残基是RIPK3诱导磷酸化的位点。当磷酸化的RIPK3(p-RIPK3)与MLKL的4HBD结 合,引 起MLKL磷酸化(p-MLKL),从而使MLKL从无活性的单体结构转变为有活性的寡聚体结构。p-MLKL与p-RIPKs结合形成坏死小体,导致p-MLKL向质膜迁移,并与质膜上的磷脂酰肌醇脂结合,锚定在细胞的质膜上,破坏膜的结构和功能的完整性,促使细胞发生死亡[14]。p-MLKL锚定于细胞器线粒体膜,导致线粒体膜电位去极化和线粒体功能受损。p-MLKL锚定于细胞膜时,促进胞外Ca2+和Na+内流,增加细胞膜对水的通透性,引起细胞肿胀、崩解和 死 亡[8,15]。研 究 发 现,使 用 选 择 性RIPK1或MLKL磷 酸化抑制剂,抑 制MLKL或RIPK3蛋 白的表达均可抑制程序性细胞坏死的进程[16]。这些研究结果说明了RIPK3及MLKL分别发挥了启动和执行程序性细胞坏死的作用,为确定参与程序性细胞坏死的调节因子提供了直接的科学证据。
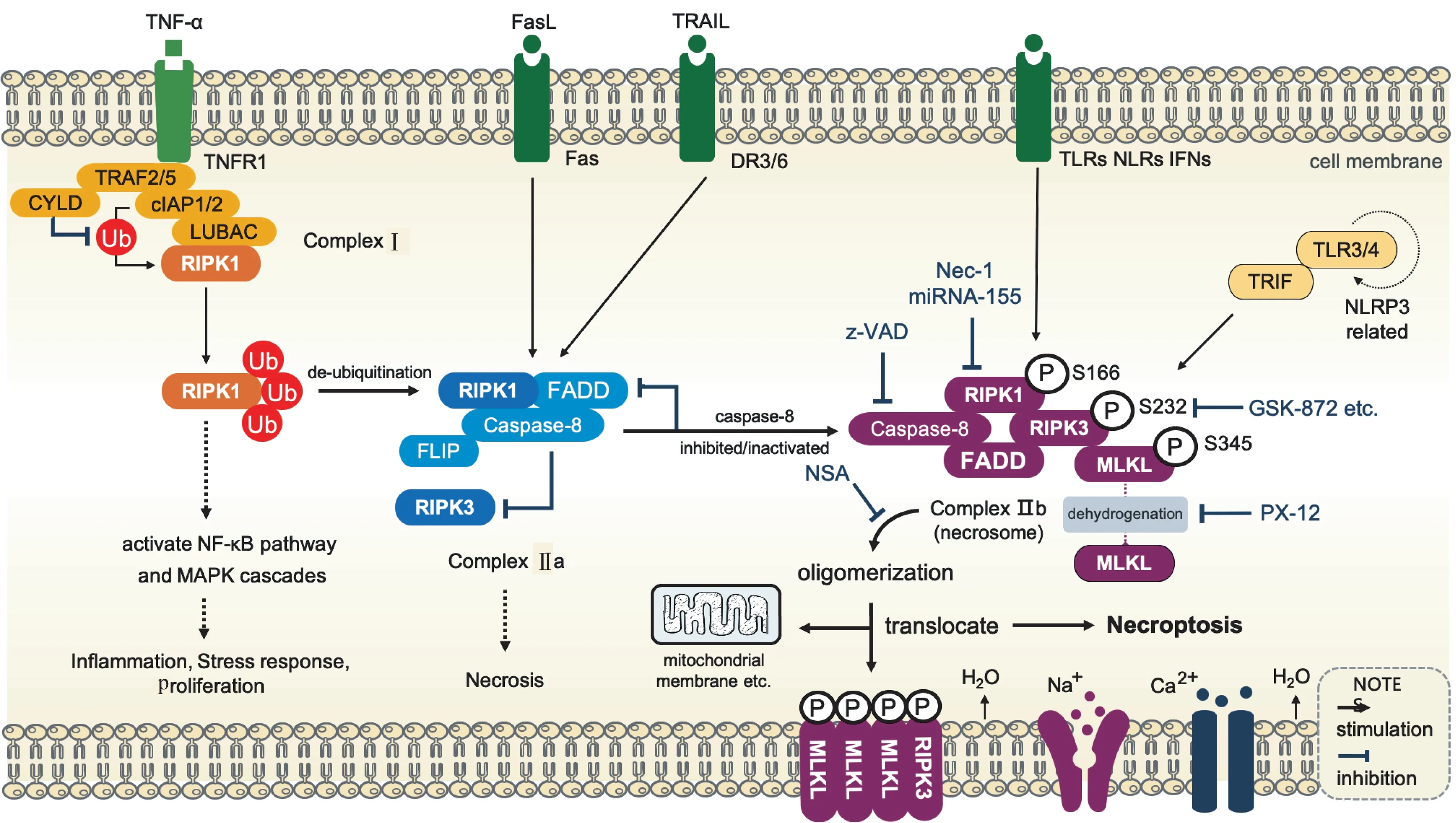
图1 程序性细胞坏死的分子信号调节通路及抑制剂作用机制的示意图Fig 1 Chematic diagram of molecular signaling pathways and inhibitor mechanisms in necroptosis
RIPK1/RIPK3的磷酸化蛋白质的磷酸化对激酶活性有重要影响。RIPK是一类含有高度保守的丝氨酸/苏氨酸激酶结构域的蛋白激酶[17]。RIPK1的C端含死亡结构域和RIP同源结合基序(RIP homotypic interaction motif,RHIM)结构域[18-19]。RIPK1和RIPK3的N端含有多个磷酸化位点的结构域。其中,RIPK1的Ser89和Ser161磷酸化可激活程序性细胞坏死的过程。体外激酶分析实验表明,RIPK1的Ser166位点能够发生自磷酸化[20],并激活下游IL-1β的合成,这被认为是介导程序性细胞坏死的一个关键起始位点。
在RIPK的序列中RIHM的结构域和RIPK3磷酸化的活性区是激活程序性细胞坏死信号通路的必要条件。RIPK3的Ser232是重要的磷酸化位点[8]。模拟磷酸化研究显示,RIPK3的Ser232磷酸化激活RIPK3激酶的活性,通过RIPK3的Ser227募集和结合MLKL[21],从而执行细胞坏死的指令。RIPK3过表达也能促进程序性细胞坏死的发生[22],这提示RIPK3在程序性细胞坏死发生中的重要性。
研究程序性细胞坏死的常用方法在人们了解蛋白质磷酸化是程序性细胞坏死启动的关键环节以前,对程序性细胞坏死的判断通常采用细胞形态学结合药物抑制剂的间接方法来判断。例如,根据形态学,人们将几种细胞死亡方式(凋亡、坏死、自噬)相比较,发现了程序性细胞坏死特有的形态表现,包括胞膜破裂、细胞内容物直接释放、DNA的逸出和双链断裂等,药物抑制剂可以抑制这种坏死细胞数。由此,研究者间接判断为该坏死细胞存在程序性细胞坏死的现象。随着研究的深入,人们对程序性坏死机制有了更深刻的了解,先后发展和制备了特异性抗体和不同环节的工具药。目前,科学家已经建立了形态学、功能学和生化分析等多种手段供相关研究。
形态学分析方法已知,原位末端转移酶标记法(TdT-mediated dUTP nick end labeling,TUNEL)可以标记双链DNA断裂,TUNEL阳性标记的细胞可以反映存在凋亡或坏死。在这个基础上,人们使用药物拮抗剂抑制RIPK蛋白表达和磷酸化,再结合TUNEL标记技术,在显微镜下观察TUNEL标记细胞数量有无变化。若使用RIPK抑制剂可以减少TUNEL标记细胞数,就可以明确该TUNEL标记细胞与程序性细胞坏死有关。Degterev等[3]采用该技术研究报道了RIPK1抑制剂Nacrostatin-1(Nec-1)能够拮抗程序性细胞坏死进程,从而在脑缺血损伤的病理过程中产生保护作用,还明确了RIPK1介导程序性细胞坏死的过程。
随着对激酶RIPK1/RIPK3结构以及活性机制的深入了解,人们发现该类蛋白磷酸化是程序性细胞坏死发生的核心环节。采用选择性磷酸化的RIPK1/3及磷酸化的MLKL抗体结合免疫荧光多标记组织化学染色技术,我们能够在共聚焦荧光显微镜下直观而清晰地观察到细胞的存亡与细胞内特定磷酸化蛋白的定位关系。这些技术的发明为了解组织中存在程序性细胞坏死提供了直接的分析手段。
免疫印迹法蛋白质印迹法(Western blot,WB)也被用于开展程序性细胞坏死机制的研究。根据磷酸化和非磷酸化蛋白的相对分子质量不同,电泳将磷酸化和非磷酸化蛋白分离,结合采用特异性磷酸化的RIPK1/3及MLKL抑制剂,通过分析组织样品中不同相对分子质量的RIPK1/3及MLKL差异条带的变化,推测该通路的被激活的状态。研究者们也可以选用针对不同的磷酸化位点的RIPK1/3及MLKL抗体进行WB分析。RIPK1/3及MLKL磷酸化位点的发现为研发各类分子的拮抗剂、小分子干扰剂和抗体提供了重要理论基础。
药物抑制剂的分析RIPK1/3和MLKL的磷酸化是激活程序性细胞坏死的信号通路的必要条件。据此,已研发了多种针对程序性坏死信号通路不同环节的抑制剂。其中,RIPK-1抑制剂Nec-1[3]和MLKL抑制剂NSA[23]是研究较为深入的两种。另外,Nec-7也能抑制程序性细胞坏死[24],但其作用机制尚不明确。NSA在细胞实验中表现出对MLKL有较强抑制作用。在整体应用时,NSA的代谢极快,从而限制了全身应用的有效性。研究发现,一种靶向治疗晚期黑色素瘤的药物Tafinlar具有抑制MLKL的Ser358磷酸化、阻止MLKL被募集的作用[25]。另有研究报道,miRNA-155[26]亦可通过靶向阻断RIPK1合成,抑制细胞坏死的发生。GSK系列的小分子具有抑制RIPK3激酶活性的作用,Pazopanib是一种新颖的酪氨酸激酶抑制剂,能抑制RIPK1/3磷酸化,从而抑制酶活性。实验研究证明,RIPK3激酶抑制剂具有靶向抗肿瘤血管生成作用,其是否具有临床治疗效果依然需要进一步研究[27]。各药物抑制剂及其作用机制详见表1。
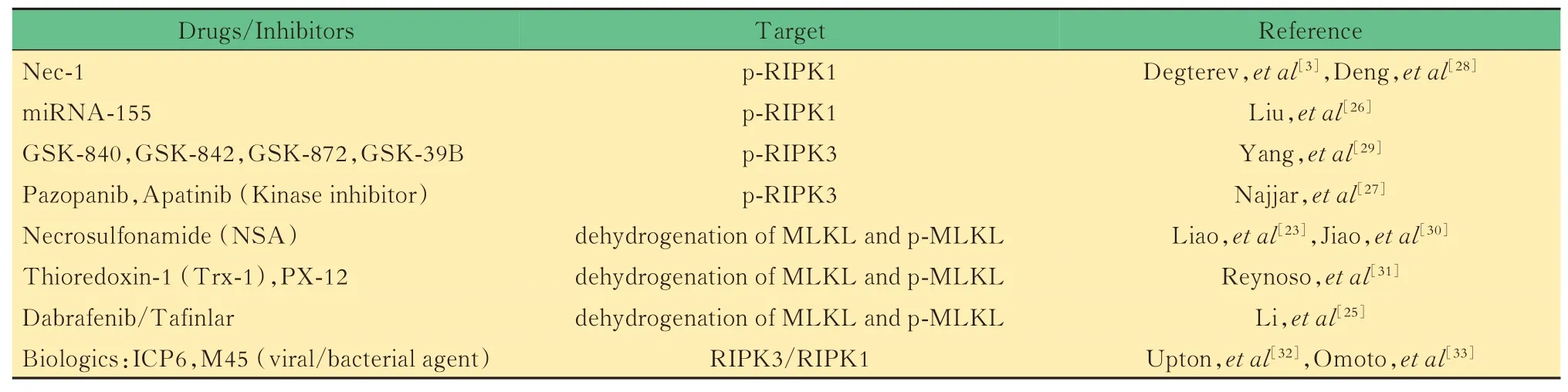
表1 程序性细胞坏死的药物抑制剂及其作用机制Tab 1 Drug inhibitors of necroptosis and their mechanism of action
回顾开展程序性细胞坏死研究的技术发展历程,从早期的药物分析法到采用特异性抗体进行形态学和生化的蛋白磷酸化分析法,反映了从间接推测到直接提供依据的研究发展的过程。这些技术的发展,对深入研究程序性细胞坏死的病理生理学意义提供了重要的分析工具。
程序性细胞坏死与脑内非感染性炎症程序性细胞坏死发生时,由于细胞膜的破坏,细胞内容物从胞质中释放,这些内源性分子属于损伤关联分子 模 式(damage associated molecular patterns,DAMPs),免疫细胞能够通过模式识别受体摄取这些分子,从而引起剧烈的炎症反应。2012年,有研究发现炎症小体NOD样受体蛋白3(Nod-like receptor pyrin domain-containing protein 3,NLRP3)或caspase-8能在RIPK3的作用下激活炎症小体的形成[34]。当然,RIPK1和MLKL同样也参与到了炎症反应中,其各自的作用机制仍有待进一步阐明。
经典的NLRP3炎症小体活化由两种信号共同激活:第一信号激活TLR4信号通路,促进细胞核因子-κB(nuclear factor kappa-B,NF-κB)激活白介素-1β(interleukin-1β,IL-1β)和IL-18等前体产生;第二信号则促进NLRP3/ASC/pro-caspase-1蛋白复合物炎症小体的形成,其中pro-caspase-1活化形成caspase-1,后者将IL-1β和IL-18前体剪切活化并释放到胞外[35]。非经典的活化则不依赖TLR4信号通路,由caspase-4/5/11介导发生,在拮抗和清除病原微生物感染中发挥重要作用[34]。
整体脑缺血实验和离体培养细胞的实验研究均证明NLRP3炎症小体参与缺血缺氧诱导的神经元死亡过程,NLRP3和ASC表达增加,以及caspase-1活化,下游炎症因子IL-1β、IL-6均参与了OGD诱导的小鼠原代皮层神经元的坏死过程[36]。另外,脑缺血后坏死的神经元释放大量TNF-α和IL-1β,引发邻近小胶质细胞向M1方向极化[37],加剧促炎因子和毒性物质释放,导致神经细胞死亡。因此被认为炎症反应同样是缺血性脑损伤预后不良的因素。
程序性细胞坏死与缺血性脑损伤近年来研究报道了RIPKs-MLKL信号通路激活参与缺血性损伤脑内神经元的细胞程序性坏死作用。采用免疫荧光染色和蛋白质磷酸化分析发现,缺血损伤脑内表达p-RIPK1和p-MLKL蛋白含量增加,采用RIPK1抑制剂Nec-1能够阻止缺血损伤引起的脑内p-RIPK1以及p-MLKL蛋白表达,减少p-RIPKTUNEL双标记的程序性坏死细胞数,缩小梗死面积,促进神经功能的恢复[28]。另外,离体实验发现小分子化合物GSK-872是一种高效和特异的RIPK3抑制剂。在离体的氧糖剥夺(oxygen glucose deprivation,OGD)模型和整体脑缺血模型上均证明,GSK-872具有抑制低氧诱导RIPK1/3和MLKL蛋白磷酸化的作用,同时减少神经元的死亡和减轻脑损伤的程度,从而发挥神经保护作用[29]。进一步的机制分析发现,用GSK-872或siRNA抑制RIPK3的表达,可以抑制由OGD诱导的RIPK1/3和MLKL蛋白磷酸化以及缺血缺氧诱导的脑内低氧诱 导因子-1α(hypoxia inducible factor-1α,HIF-1α)的表达。但是,采用siRNA抑制HIF-1α的表达可产生神经保护效应,却不影响RIPK1/3和MLKL蛋白及其磷酸化蛋白的表达量。由此提示缺血缺氧激活脑内程序性坏死信号分子RIPK1/3和MLKL磷酸化,后者可能是通过诱导HIF-1α表达产生细胞死亡。
程序性细胞坏死与神经退行性疾病程序性细胞坏死最先被发现于脑缺血损伤的病理表现中。越来越多的研究报道,程序性细胞坏死参与到多种神经退行性疾病的病理进程中,包括帕金森病(Parkinson’s disease,PD)[38]、肌 萎 缩 侧 束 硬 化(amyotrophic lateral sclerosis,ALS)[39-40]、多 发 性 硬化(multiple scalerosis,MS)[41]和 阿 尔 茨 海 默 病(Alzheimer’s disease,AD)[42]。这类疾病的病理特征均表现为神经细胞进行性丢失和死亡的过程。目前,我们对这类疾病的病理机制了解还不甚清楚,但是近年的研究证明,程序性神经细胞坏死参与这些神经退行性疾病的发生和发展过程(表2)。

表2 程序性细胞坏死与神经疾病的关系Tab 2 Relationships between necroptosis and neurological diseases
MS是一种常见的中枢神经系统退行性疾病,其病理特征为神经元轴突的少突胶质细胞的丢失和脱髓鞘。在动物实验模型研究中已发现,在MS发生发展过程中,TNF-α作为促炎细胞因子,在caspase-8缺乏的情况下,激活RIPK1和RIPK3介导的程序性细胞坏死通路,引起神经元的退行性病变,诱导少突胶质细胞的变性死亡。抑制RIPK1可防止TNF-α诱导的少突胶质细胞死亡,表明程序性坏死参与MS病理的发生发展过程,靶向RIPK1抑制剂可能是MS的一种治疗策略。
ALS是以脊髓运动神经元丢失,导致轴突溃变为主的病变,患者表现为进行性运动功能退变,晚期患者表现为偏瘫和呼吸困难,乃至窒息。因此,该病俗称“渐冻症”。ALS的病理机制并不清楚,有报道ALS分为家族性和散发性。目前认为SOD缺失导致氧化损伤是诱导ALS发生的主要原因之一。因此,SOD1G93A转基因小鼠往往用于ALS疾病模型研究。另外,还有些基因突变参与疾病发生,如视神经蛋白(optineurin,OPTN)缺失。Ito等[39]研究表明,在SOD1G93A转基因小鼠和人类ALS患者的病理样本中,通常可检测到RIPK1和RIPK3。另外,OPTN缺失转基因小鼠表现为脊髓运动神经元死亡和轴突溃变。该作用通过Nec-1抑制RIPK1活性而减弱。敲除RIPK3的表达也可以阻止OPTN缺失小鼠引起的脊髓运动神经元丢失和轴突溃变。由此提示,在ALS的病理发展过程中,脊髓神经组织内的RIPK1、RIPK3和MLKL信号通路激活并参与进行性运动神经元死亡和神经轴突的退化。此外,抑制RIPK1或RIPK3激酶能否成为这类疾病的临床治疗药物还有待深入研究。
PD是一种以脑内黑质多巴胺神经元丢失为病理特征的,精神行为异常和运动功能减弱为临床表现的神经退行性疾病。引起多巴胺神经元死亡的机制十分复杂,包括神经元自身代谢性氧化损伤、疾病基因和炎症反应等。近年来的研究提示程序性细胞坏死参与多巴胺神经元死亡的病理过程。PD研究的常用细胞模型由药物或基因突变诱导制备。其中,6-羟基多巴胺(6-hydroxydopamine,6-OHDA)是常用的工具药。研究者利用6-OHDA诱导培养细胞死亡模型发现,6-OHDA引起线粒体功能障碍,导致自噬增加,组织蛋白酶B(cathepsin B)表达增加,Bcl-2表达减少,引起细胞死亡。而RIPK1的抑制剂Nec-1能预防6-OHDA的细胞毒性,稳定线粒体的膜电位,抑制过度的自噬,同时降低微管相关蛋白轻链3(microtubule-associated protein 1 light chain 3,LC3)和组织蛋白酶B的表达,增加了Bcl-2的表达,减少细胞的死亡[38],由此提示了RIPK1磷酸化激活参与帕金森的病理性细胞死亡过程。
AD是以脑内胆碱能神经元丢失、老年斑和神经纤维缠结形成为病理特征,临床主要表现为精神活动异常和认知功能减退为主的神经退行性疾病。主流观点认为引起AD病理变化的机制为脑内Aβ的聚集形成老年斑,Tau蛋白异常磷酸化引发神经纤维缠结。老年斑和神经纤维缠结形成是进一步触发脑内神经元病理发展的元凶。因此,科学家建立了多种与病理机制有关的模式动物模型开展研究,包括APP/PS1和5xFAD小鼠。最近,神经炎症和脑内的免疫反应参与AD病理过程受到广泛重视和认可。小胶质细胞被认为是脑内的免疫细胞,它介导脑内炎症和免疫反应。脑内Aβ沉积可诱导小胶质细胞向M1方向极化,促进炎症小体的形成[43]。已知炎症小体具有激活RIPK1/3的作用,从而诱导程序性细胞坏死的发生。为了证明炎症介导的程序性细胞坏死参与AD脑内神经元退行性病理过程,Caccamo等[42]首先观察AD患者脑内表达程序性细胞坏死标志蛋白RIPK 1和MLKL;其次,采用两种常用的AD模式动物(APP/PS1和5xFAD小鼠)进行整体动物和离体神经元培养模型开展研究。结果发现,在AD患者脑内表达程序性坏死标记蛋白RIPK1和MLKL的量增加,这种增加与脑内神经元的存活以及认知功能呈负相关;在5xFAD小鼠脑内观察到RIPK1和MLKL表达增高的类似结果。同时,APP/PS1小鼠脑内高表达MLKL蛋白导致海马神经元丢失,伴有学习能力和记忆功能减退症状。反过来,采用Nec-1s抑制RIPK/MLKL信号通路活性,具有增加APP/PS1小鼠皮层的培养神经元存活的作用。若Nec-1s全身应用3周后,5xFAD小鼠脑内p-MLKL表达下降,Fluoro-Jade着色的退行性病变神经元数量明显减少。以上结果明确提示减少脑内RIPK/MLKL信号通路活性具有减少AD转基因动物脑内神经元死亡和改善认知功能的作用[42]。这些证据有力地说明程序性细胞坏死参与了AD脑内神经元丢失的病理过程。这些结果不仅拓宽了人们对AD病理机制的理解和治疗研究的思路,还提示通过药物有效减少程序新细胞坏死可能有助于延缓疾病的症状发生和发展。
结语程序性细胞坏死是免疫炎症相关的一种细胞死亡形式,其分子机制对胚胎发育和新生儿成长有重要影响。同时,程序性细胞坏死也参与到组织稳态调节和各种病理过程的发展中。免疫炎症反应涵盖了诸多复杂的机制,是由免疫系统中各种因素共同制衡而产生的。程序性细胞坏死在不同时期、不同部位影响着免疫系统,探索其如何调控免疫细胞的分子表型能够帮助我们加深理解,并有助于区分各类细胞死亡的形式及其病理生理学意义。此外,RIPK1/RIPK3的靶向研究对开展神经退行性疾病的病理机制和防治策略方面的研究有重要意义,未来研究的焦点在于深入相关分子作用机制,并致力于研发调控程序性细胞坏死的药物。
作者贡献声明钱奕茗论文构思、撰写和修订,绘制图表。孙凤艳论文审校和修订。
利益冲突声明所有作者均声明不存在利益冲突。
