Synthesis of S-monofluoromethyl phosphorothioates from PV-H compounds and PhSO2SCH2F
2021-04-02RenyiPngRuichoYoShiyoLuYichengZhouWenboChen
Renyi Png,Ruicho Yo,Shiyo Lu,Yicheng Zhou,Wenbo Chen,b,*
a Shanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, Shanghai University of Electric Power, Shanghai 200090, China
b CAS Key Laboratory of Energy Regulation Materials, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
ABSTRACT S-Monofluoromethyl phosphorothioates represent an important class of organofluorine compounds and are reported here for the first time.A series of S-monofluoromethyl phosphorothioates are conveniently synthesized from different PV-H compounds and PhSO2SCH2F under mild conditions.The method is compatible with common functional groups and provides potential opportunities to synthesize new bioactive molecules for medicinal chemistry.
Keywords:Fluorine Monofluoromethylthio Phosphorothioates PV-H compounds PhSO2SCH2F
In the last few dec ades, fluorine-containing compounds have received considerable interest in the fields of life science and materials science due to unique “fluorine effect” [1-7].The introduction of the fluorine atom or fluorinated group into the specific position of the molecules has been a powerful method to tune the properties of the functional molecules [8-16].Recently,fluoroalkylthio groups, such as trifluoromethylthio (-SCF3)[17-23], difluoromethylthio (-SCF2H) [24-31], monofluoromethylthio (-SCH2F) [32,33] have drawn more attention from medicinal chemists owing to their high lipophilicity and metabolic stability.Various reagents and methods have been developed to prepare fluoroalkylthiolated compounds[34,35].Compared to the extensive reports on trifluoromethylthio (-SCF3) and difluoromethylthio (-SCF2H) compounds, the studies on the monofluoromethylthio(-SCH2F)compounds are very limited because of the lack of efficient monofluoromethylthiolating reagents and methods.The known monofluoromethylthio compounds have demonstrated their huge potential in pharmaceutical industry, evidenced by the anti-inflammatory drug fluticasone propionate [36] and nonsteroidal antiflammatory agent cathepsin K inhibitors [37](Fig.1a).Therefore, it is an urgent task to prepare diverse monofluoromethylthio compounds to meet increasing demand for medicinal chemistry.
However, to date, the reports on monofluoromethylthio compounds are mainly limited to monofluoromethyl thioethers[38] and monofluoromethyl carbothioates (Figs.1b and c).To the best of our knowledge, S-monofluoromethyl phosphorothioates are not reported yet, even they represent an important class of monofluoromethylthio compounds.Phosphorothioates have been used to substitute the native phosphate backbones to increase metabolic stability and cellular uptake in antisense oligonucleotide therapeutics [39,40].Thus, the S-fluoroalkylphosphorothioates might provide new opportunities for the development of new bioactive molecules in virtue of the advantage of fluorine.Actually,S-fluoroalkyl phosphorothioates were scarce and only S-trifluoromethyl phosphorothioates were reported in the literatures[41-43].However, the S-trifluoromethyl phosphorothioates were thermally labile and prone to lose SCF2to generate fluorophosphates, which limited their practical applications.Herein, we report a simple and efficient method to synthesize S-monofluoromethyl phosphorothioates from PV-H compounds and PhSO2SCH2F under mild conditions (Fig.1d).
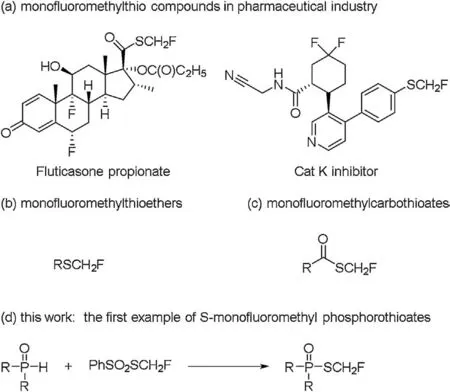
Fig.1.The approach for the preparation of S-monofluoromethyl phosphorothioates.
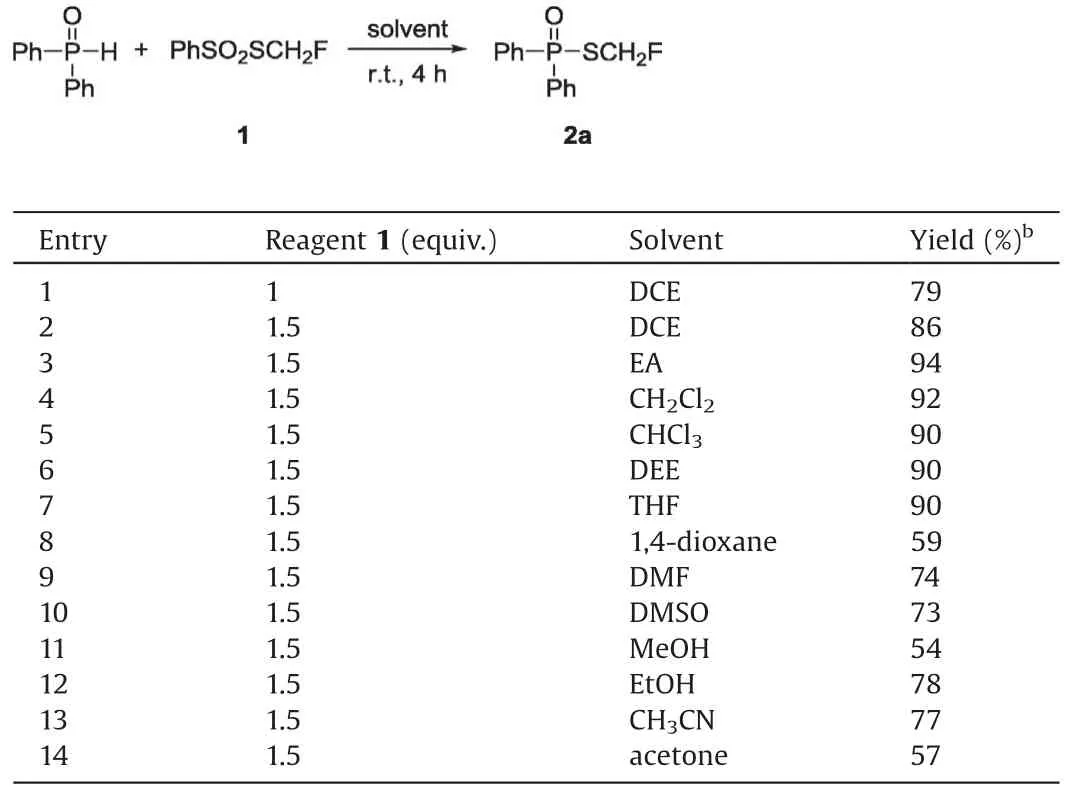
Table 1 Optimization of the reaction conditions for diphenylphosphineoxide.a
We firstly chose commercially available diphenylphosphine oxide and monofluoromethylthiolating reagent (PhSO2SCH2F) 1,which was developed by Shen and Lu[44],as the model substrates to optimize the reaction conditions (Table 1).The initial reaction condition was set at room temperature for 4 h.To our delight,the reaction occurred smoothly in 1,2-dichloroethane without additives and afforded the corresponding monofluoromethylthiolated product 2a in 79% isolated yield (Table 1, entry 1).When the amount of PhSO2CH2F was increased to 1.5 equiv.,the reaction gave full conversion of diphenylphosphine oxide and the yield of product was increased to 86% (Table 1, entry 2).Further solvent screening showed that the reaction could work in common organic solvents.The reaction in ethylacetate afforded the desired product in highest 94% yield (Table 1, entry 3).Other solvents such as dichloromethane,chloroform,diethyl ether,tetrahydrofuran were also very effective for this reaction,while more polar solvents such as 1,4-dioxane, DMF, DMSO, CH3OH, C2H5OH, CH3CN and acetone gave the product in lower yield (Table 1, entries 4-14).With the optimal conditions in hand,we expanded the scope of substrates.As shown in Scheme 1, both electron-rich and electron-deficient diphenylphosphine oxides reacted smoothly to afford the corresponding products in good yields (2a-2d).
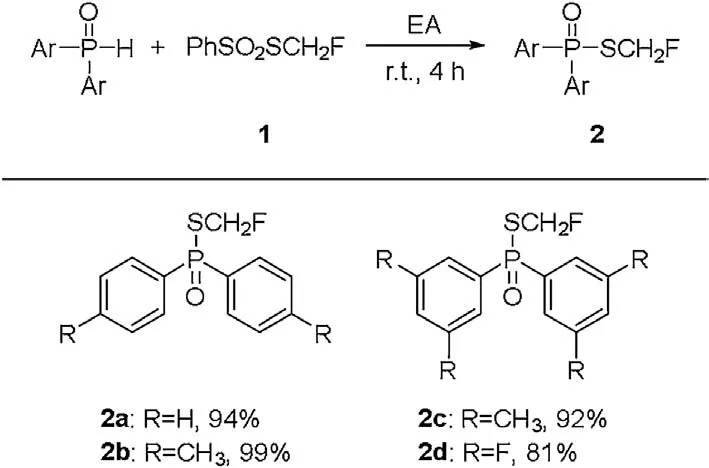
Scheme 1.Scope of monofluoromethylthiolation of diarylphosphine oxides.Reaction conditions: diaryphosphine oxides (0.50 mmol), reagent 1 (0.75 mmol)in ethyl acetate(5.0 mL),at room temperature for 4 h, under air.All yields given are isolated yields.
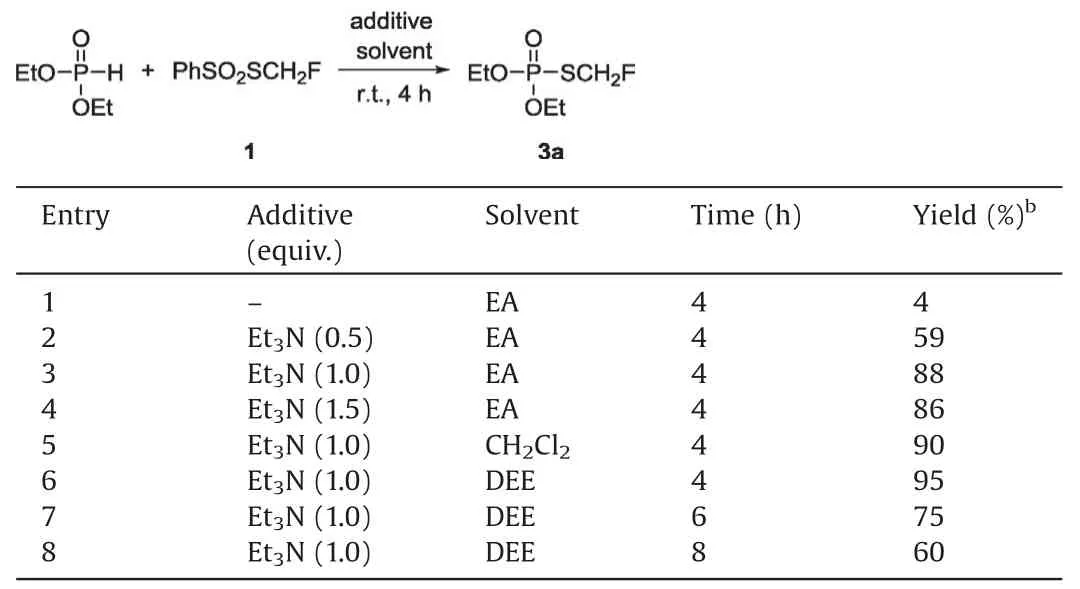
Table 2 Optimization of the reaction conditions for diethyl phosphonate.a
Subsequently, we turned our attention to the reaction of the H-phosphonate with reagent 1 to prepare diverse S-monofluoro-methyl phosphorothioates.Surprisingly, under the same conditions,the reaction of diethyl phosphonate with reagent 1 was sluggish and only gave the product 3a in 4% yield (Table 2,entry 1).With the addition of triethylamine, this reaction was significantly promoted and afforded the product in 59% yield(Table 2,entry 2).The yield was increased to 88%when 1.0 equiv.of triethylamine was added (Table 2, entry 3) and the excess of triethylamine did not help to improve the yield(Table 2,entry 4).The solvents and reaction time were also quickly screened and it was found that the reaction in diethyl ether gave the highest 95%yield (Table 2, entry 6), whereas the longer reaction time will decrease the yield (Table 2, entries 7 and 8).
Based on the optimized conditions in entry 6 of Table 2,we next investigated the substrate scope of this protocol (Scheme 2).In general, dialkyl/monoalkyl/diaryl H-phosphonates were applicable and gave the products in moderate and excellent yields.Specifically, hindered H-phosphonate such as diisopropyl phosphonate, diisobutyl phosphonate and dibenzyl phosphonate also reacted smoothly to give the products in 86%, 95% and 93% yield,respectively (3c, 3e and 3h).Nevertheless, more hindered ditertbutyl phosphonate did not react with reagent 1 under the standard condition.Furthermore,diarylphosphonate,for example,diphenylphosphonate also reacted to produce the corresponding O,O-diphenyl S-monofluoromethyl phosphorothioates product in 74% yield (3g).In addition, the reaction of monoalkyl phenyphosphonates with reagent 1 gave the full conversion within 1 h and afforded the desired product in good yields(3m-3o),which is faster than that of dialkyl phosphonates.Due to mild conditions,the common functional groups such as,bromide(3i),alkenyl(3j),cycloalkyl(3k)and thienyl(3l)were well-tolerated in this reaction.
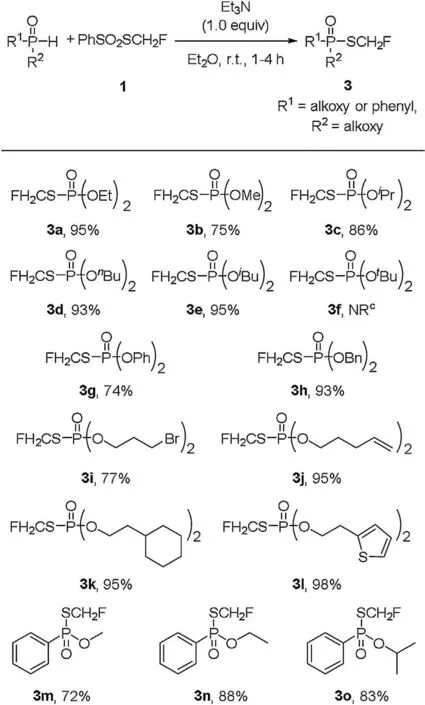
Scheme 2.Scope of monofluoromethylthiolation of H-phosphonate.H-Phosphonate (0.5 mmol), 1 (0.75 mmol) and Et3N (0.5 mmol) in diethyl ether (5.0 mL), at room temperature for 1-4 h, under air.All yields given are isolated yields.
Reagent 1 was previously reported as a radical (·SCH2F)precursor in the monofluoromethylthiolation reaction of aldehyde[45]and the addition reaction of unactivated alkene with reagent 1[44].To further explore the reaction mechanism whether this reaction involved the radical pathway, we conducted a series of controlled experiments.Two free radical trapping agents,2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and dibutylhydroxy-toluene(BHT)were added to the standard reaction of PhSO2SCH2F withdiethyl phosphonate.It was found that the two reactions were largely retarded and gave the product in 13% and 12% yield respectively, suggesting that the radical wasinvolved in the transformation (Table 3, entries 2 and 3).Furthermore,thereactionof PhSO2SCH2F with diethyl phosphonate was partially inhibited by the addition of 1.0 equiv.of a single-electron-transfer(SET) inhibitor 1,4-dinitrobenzene and 1,3-dinitrobenzene under standard conditions (Table 3, entries 4 and 5).Since this reaction occurred without the addition of the radical initiator, a single electron transfer process might be involved, where diethyl phosphonate itself acted as electron donor and initiated this reaction.

Table 3 Control experiments of reagent 1 with diethyl phosphonate.a
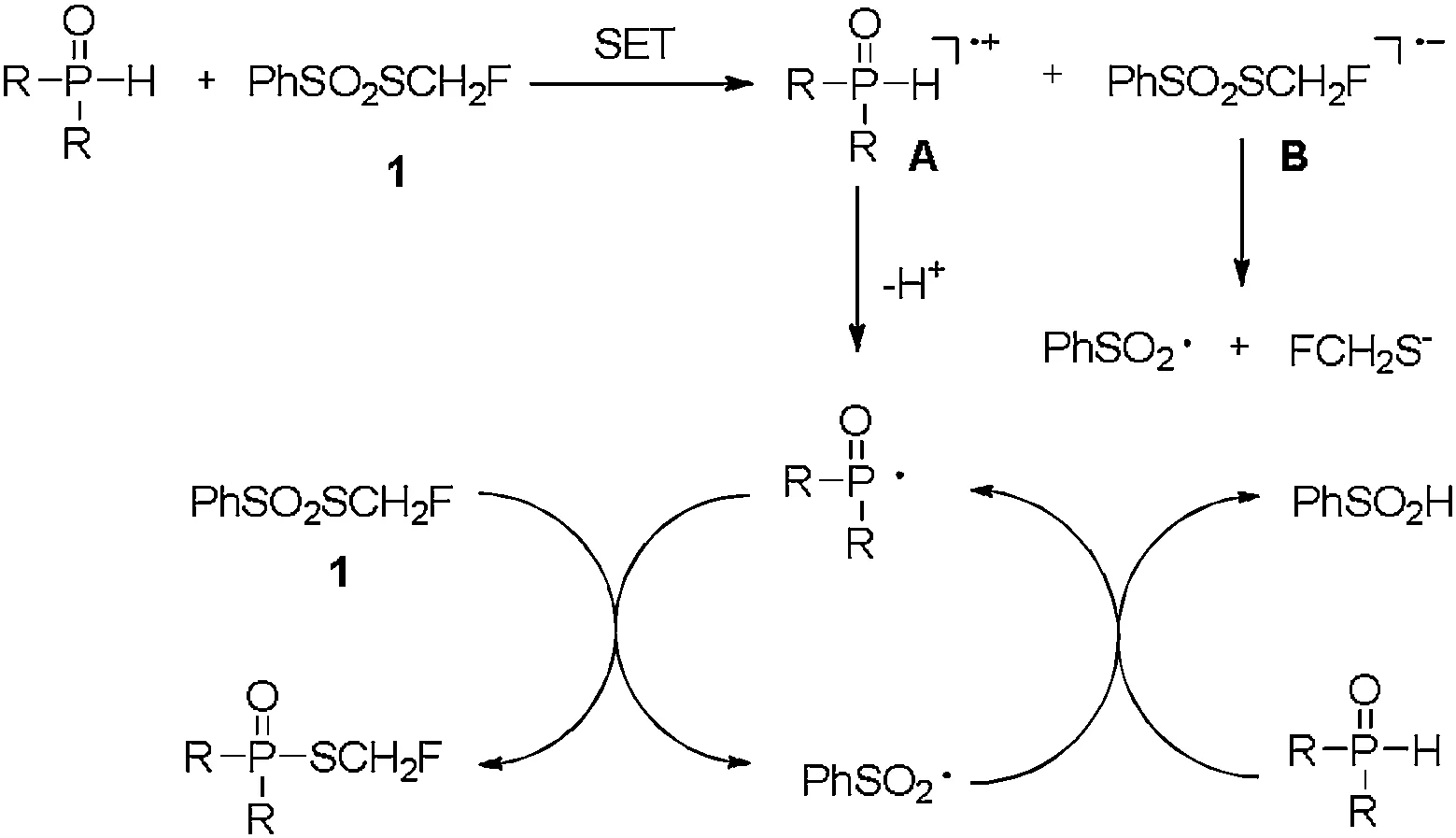
Scheme 3.Plausible mechanism.
Based on these experiments and the related literatures[24,32,45-47], we proposed a tentative mechanism for the reaction of PV-H compounds with reagent 1 (PhSO2SCH2F)(Scheme 3).In the reaction, PV-H compound donated a single electron to PhSO2SCH2F to form the radical cation A and radical anion B.Then the resulting radical cation A loses proton to generate phosphoryl radical, meanwhile, anion B decomposes to PhSO2radical and monofluoromethylthiol anion.The phosphoryl radical reacts with PhSO2SCH2F to give the corresponding Smonofluoromethyl phosphorothioates, together with the formation of PhSO2radical [44].The generated PhSO2radical might abstract the hydrogen of PV-H compound to produce the phosphoryl radical and PhSO2H to finish the radical chain cycle.More details about the mechanism of the reaction are ongoing.
In conclusion, S-monofluoromethyl phosphorothioates, as an important class of monofluoromethylthio compounds, are reported for the first time.S-Monofluoromethyl phosphorothioates were synthesized from monofluoromethylthiolated reagent PhSO2SCH2F and different PV-H compounds including diarylphosphine oxides and H-phosphonate.Due to the mild conditions, this reaction is compatible with common function groups.Preliminary mechanistic studies suggest this reaction might proceed via a single-electron-transfer pathway.This method provides potential opportunities to synthesize new bioactive molecules for medicinal chemistry .
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Natural Science Foundation of Shanghai (No.17ZR1447100), and the Science and Technology Commission of Shanghai Municipality (No.19DZ2271100).
Appendix A.Supplementary data
Supplementarymaterialrelatedtothisarticlecanbefound,inthe online version,at doi:https://doi.org/10.1016/j.cclet.2020.05.032.
杂志排行

Chinese Chemical Letters的其它文章
- A biomass based photonic crystal made of “konjac tofu”
- Hydrothermal-assisted grinding route for WS2 quantum dots (QDs)from nanosheets with preferable tribological performance
- Superiority of poly(L-lactic acid) microspheres as dermal fillers
- Zwitterionic comb-like lipid polymers encapsulating linalool for increasing the fragrance retention time
- Construction of a nano-rectangular Zn-Nd complex with near-infrared luminescent response towards metal ions
- Synthesis and structure of Au19Ag4(S-Adm)15 nanocluster:Polymorphs and optical properties