植物乳杆菌KLDS1.0391 PurR和PurL蛋白的原核表达、纯化及其与细菌素合成启动子的相互作用
2021-03-31李欣芮范小飘高文文尚佳萃赵鹏昊孟祥晨
李欣芮,赵 桉,范小飘,高文文,尚佳萃,赵鹏昊,赵 乐,周 雪,孟祥晨
(东北农业大学 乳品科学教育部重点实验室,黑龙江 哈尔滨 150030)
植物乳杆菌KLDS1.0391分离自内蒙古传统发酵乳制品,能代谢产生对革兰氏阳性菌和阴性菌都有抑制作用的细菌素[1],2% NaCl胁迫可促进其细菌素合成[2],转录组学分析表明,盐胁迫时purR基因和purL基因表达显著下调,推测其可能参与细菌素合成的调控作用。
purR基因编码的PurR蛋白是嘌呤从头生物合成途径中的阻遏蛋白,对嘌呤生物合成途径中11 个pur系列的结构基因具有转录调节作用。不同细菌中pur基因的分布和调控遵循不同的规则[3],在革兰氏阴性菌大肠杆菌中,嘌呤生物合成基因散布在染色体各处,在革兰氏阳性菌枯草芽孢杆菌中,所有参与嘌呤合成的基因都分布在一个转录单元,即嘌呤操纵子[4]。关于植物乳杆菌purR的研究较少,但有研究表明乳酸乳球菌中的purR与枯草芽孢杆菌中的purR同源[5]。许多代谢调节蛋白具有双重功能甚至是未被发现的功能,purR基因除对嘌呤生物合成具有重要的调控作用外,还对嘧啶合成途径中的pyrC与pyrD基因、与Cysosine转运吸收有关的codBA基因、编码5-磷酸核糖焦磷酸合成酶的prs基因以及甘氨酸合成代谢和一碳单位合成的glyA基因和gcv操纵子等具有调控作用[6-7]。除此之外,近年研究表明,金黄色葡萄球菌中purR突变增强了其致病力[8],进一步研究发现,purR突变体表现出的毒力增加与嘌呤合成无关,PurR能够结合并调节嘌呤生物合成途径之外的启动子元件,作为一种转录因子,直接结合至毒力基因的启动子,调节毒力相关基因的表达[9]。因此,PurR可能存在尚未被开发的功能。
purL基因也参与嘌呤生物合成途径,对于腺嘌呤和硫胺素的生物合成是必需的。Hava等[10]已经证明其是肺炎链球菌中重要的毒力因子;Xia Yanfei等[11]研究发现由purL基因调控的嘌呤生物合成中间体可以影响枯草芽孢杆菌OKB105菌株的杀虫活性;Alcantara等[12]发现purL基因突变会显著降低流产布鲁氏菌(Brucella abortus)的毒性;Maegawa等[13]报道了艰难梭菌(Clostridium difficile)毒素的产生与PurL底物的积累呈反比;Ge Xiuchun等[14]也发现purL基因与链球菌的生物膜形成有关;Liu Quanli等[15]还发现purL基因突变的解淀粉芽孢杆菌(Bacillus amyloliquefaciens),其细菌素Subltilosin A的产量增加显著,抑菌能力增强。可见,purL基因与微生物的生理活动有密切关系。
植物乳杆菌KLDS1.0391全基因组测序以及BLAST分析发现[16],该菌包含4 个与细菌素合成相关的操纵子plnEFI、plnBD、plnGHSTUVW及plnXY,不同植物乳杆菌pln基因簇结构相似但不完全相同,但是所有的pln基因簇都具有非常相似的启动子,每个操纵子的启动子由2 个半保守的直接重复序列组成[17],通过全基因组测序结果可以比对获得植物乳杆菌KLDS1.0391中的plnE和plnG启动子[18]。
用于研究启动子与调控蛋白相互作用的常用方法有凝胶阻滞实验(electrophoretic mobility shift assay,EMSA)[19-20]、DNase I足迹法[21-22]、染色质免疫沉淀技术[23-25]以及近几年发展起来的生物膜干涉(bio-layer interferometry,BLI)技术[26-27]等,能够检测启动子核酸靶标与蛋白相互作用以及检测二者之间的结合以及解离常数等。
本研究主要目的是通过体外分子相互作用的方法探究PurR和PurL蛋白能否与植物乳杆菌KLDS1.0391启动子相结合,进而判断这两个蛋白是否直接参与细菌素合成的调控。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
植物乳杆菌KLDS1.0391为东北农业大学乳品科学教育部重点实验室保藏;pMD™19-T克隆载体、大肠杆菌JM109感受态细胞 宝日医生物技术(北京)有限公司;pQE-30表达载体、大肠杆菌M15感受态细胞华越洋生物(北京)科技有限公司。
1.1.2 试剂与培养基
质粒小提试剂盒、细菌基因组DNA提取试剂盒、通用型DNA纯化回收试剂盒、TaqDNA Polymerase、D2000 DNA Marker 天根生化科技(北京)有限公司;PrimeSTAR®HS DNA Polymerase、DL5000 DNA Marker、DNA Ligation Kit、限制性内切酶KpnI、BamHI、HindIII 宝日医生物技术(北京)有限公司;卡那霉素(Kana)、氨苄青霉素(Amp)、异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG) 美国Amresco公司;Bradford蛋白质量浓度测定试剂盒、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)凝胶快速配制试剂盒、SDS-PAGE蛋白上样缓冲液(5×)、Annealing Buffer for DNA Oligos(5×)、EMSA/Gel-Shift结合缓冲液(5×) 上海碧云天生物技术有限公司;预染彩虹蛋白Marker(10~190 kDa) 大连美仑生物技术有限公司;HisSep Ni-NTA Agarose Resin上海翊圣生物科技有限公司;牛血清白蛋白(bovine serum albumin,BSA) 美国Sigma公司;其他试剂为国产分析纯。
LB培养基及MRS培养基配制方法参照文献[28-29]。
1.2 仪器与设备
梯度聚合酶链式反应(polymerase chain reaction,PCR)仪 德国耶拿分析仪器股份公司;DYY-10C型电泳仪 北京市六一仪器厂;凝胶成像系统 美国UVP公司;QuickDrop超微量核酸蛋白分析仪、SpectraMax i3x多功能酶标仪、Octet RED96e分子互作分析系统美谷分子仪器(上海)有限公司;超声波细胞粉碎机宁波新芝生物科技股份有限公司;蛋白纯化柱 美国Thermo Fisher公司;蛋白质电泳仪、GelDoc EZ凝胶成像系统 美国Bio-Rad公司。
1.3 方法
1.3.1 引物的设计与合成
根据植物乳杆菌KLDS1.0391全基因组测序结果得到的purR和purL的基因序列,以及表达载体pQE-30质粒所包含的酶切位点,应用软件Primer Premier 5.0设计引物。在purR基因上下游引物的5’端分别引入限制性内切酶KpnI和HindIII的酶切位点以及相应的保护碱基,purR基因上游引物(FR)为5’-GGGGTACCATGAAAGTACGTA GAAGCGAACG-3’(下划线是KpnI酶切位点),下游引物(RR)为5’-CCCAAGCTTCTAGACTTCCGCACCGAA AAC-3’(下划线是HindIII酶切位点);在purL基因上下游引物的5’端分别引入限制性内切酶BamHI和HindIII的酶切位点以及相应的保护碱基,purL基因上游引物(FL)为5’-CGCGGATCCATGCTTAAAAAGCAACCATTATC-3’(下划线是BamHI酶切位点),下游引物(RL)为5’-C CCAAGCTTTTACTTCAGTAGGCATGGTAAGG-3’(下划线是HindIII酶切位点)。引物由吉林库美生物科技有限公司合成。
1.3.2 重组克隆载体的构建与鉴定
用细菌基因组DNA提取试剂盒提取植物乳杆菌KLDS1.0391基因组DNA作为模板,进行PCR扩增。purR基因的扩增体系:DNA模板1 μL,上下游引物(10 μmol/L)各2 μL,高保真酶PrimeSTAR®HS DNA Polymerase(2.5 U/μL)0.5 μL,5×PrimeSTAR Buffer(Mg2+plus)10 μL,dNTP Mixture(各2.5 mmol/L)4 μL,dd H2O 30.5 μL,总体系50 μL。扩增过程:94 ℃预变性3 min,98 ℃变性10 s,63 ℃退火15 s,72 ℃延伸1 min,30 个循环;72 ℃延伸10 min。purL基因扩增退火温度为60 ℃,其余均与purR基因的扩增过程相同。扩增后使用通用型DNA纯化回收试剂盒回收目的基因,在目的基因3’端加A碱基后,与克隆载体pMD 19-T simple按一定比例混合,于16 ℃连接,通过热激法转入大肠杆菌JM109感受态细胞,涂布于含有Amp的平板上,提取阳性菌落的质粒进行双酶切鉴定,鉴定正确的重组子进行测序鉴定,分别命名为pMD 19-T simple-purR和pMD 19-T simple-purL。
1.3.3 重组表达载体的构建与鉴定
用质粒小提试剂盒提取坚定正确的质粒pMD 19-T simple-purR和pMD 19-T simple-purL,前者用KpnI和HindIII进行分步酶切,后者用BamHI和HindIII进行分步酶切,酶切后的产物进行琼脂糖凝胶电泳,切胶回收目的基因purR和purL。将回收的目的基因与用相同酶处理后的pQE-30质粒按一定比例混合,于16 ℃连接,通过热激法转入大肠杆菌M15感受态细胞,涂布于含有Amp以及Kana的平板上,提取阳性菌落的质粒进行双酶切鉴定,鉴定正确的重组子进行测序鉴定,分别命名为pQE-30-purR和pQE-30-purL。
1.3.4 PurR蛋白和PurL蛋白的诱导表达及鉴定
将鉴定正确的菌株pQE-30-purR和pQE-30-purL分别接种于含Amp 100 μg/mL和Kana 25 μg/mL的LB培养基中,37 ℃摇床振荡培养至菌液OD600nm达到0.6~0.8时,加入IPTG(终浓度为0.5 mmol/L)进行蛋白表达,隔2 h取菌液离心收集菌体,菌体用磷酸盐缓冲液(phosphate buffer saline,PBS)洗涤后加入1/10原体积的PBS进行重悬,加入SDS-PAGE上样缓冲液煮沸后用于SDS-PAGE分析。
1.3.5 PurR蛋白和PurL蛋白的纯化与透析
PurR蛋白的纯化与透析:按1%接种量将菌株pQE-30-purR接种至100 mL含Amp 100 μg/mL和Kana 25 μg/mL的LB培养基中,37 ℃摇床振荡培养至菌液OD600nm达到0.6~0.8时,加入IPTG(终浓度为0.5 mmol/L)继续培养6 h,8 000 r/min离心5 min收集菌体,PBS洗涤2 次后,加入10 mL裂解液(20 mmol/L Tris-HCl、0.5 mol/L NaCl、20 mmol/L咪唑,pH 7.9)进行超声破壁处理(130 W 40%的功率,超声5 s,静置5 s),直至菌液澄清不黏稠,全程冰上操作。破壁后的菌液12 000 r/min离心10 min取上清液,上清液过0.45 μm滤膜后,加入平衡好的Ni-NTA纯化柱中,轻轻搅动,室温孵育0.5~1 h,用梯度咪唑浓度洗脱液(20 mmol/L Tris-HCl、0.5 mol/L NaCl,咪唑浓度分别为20、50、100、150、200、300、500 mmol/L,pH 7.9)洗脱并回收PurR蛋白。纯化后的PurR蛋白加入准备好的透析袋中,于透析液(20 mmol/L Tris-HCl、0.5 mol/L NaCl,pH 7.9)中透析除去咪唑。
PurL蛋白的纯化与透析:收集菌体后,加入10 mL PBS重悬,进行超声破壁处理。破壁后的菌液12 000 r/min离心10 min弃上清液,沉淀用PBS洗涤2 次后,加入包涵体溶解液(20 mmol/L Tris-HCl、0.5 mol/L NaCl、8 mol/L尿素,pH 7.9)彻底溶解,过0.45 μm滤膜后,加入平衡好的Ni-NTA纯化柱中,轻轻搅动,室温孵育0.5~1 h,用梯度咪唑浓度洗脱液(20 mmol/L Tris-HCl、0.5 mol/L NaCl、8 mol/L尿素,咪唑浓度分别为20、50、100、150、200、300、500 mmol/L,pH 7.9)洗脱并回收PurL蛋白。纯化后的PurL蛋白加入准备好的透析袋中,不断降低透析液中的尿素浓度,使包涵体复性,最终透析至PBS中。
1.3.6 PurR蛋白和PurL蛋白的浓缩与定量
分别用10 kDa和30 kDa的超滤管浓缩PurR蛋白和PurL蛋白,12 000 r/min、4 ℃离心。浓缩后的蛋白用Bradford蛋白质量浓度测定试剂盒测定其浓度(标准品稀释液与蛋白缓冲溶液保持一致)。
1.3.7 BLI实验
1.3.7.1 Biotin-DNA制备
plnE启动子的序列为5’-ACATTGGTATTTGAC GTTAAGAGAACGTTTTTTTACTTTTATAATTTTT TCAACAATCTGGTAAAA-3’,plnG启动子的序列为5’-AAGCCTGATGAGGACATTTATCATAAAATTA TGTACGTTAATAGATAGTTGGCATACGATAACA TT-3’,均采用5’-biotin标记的方法。biotin-ss DNA与非标记的ss DNA均由生工生物工程(上海)股份有限公司合成,biotin-ss DNA与非标记的ss DNA退火形成biotin-ds DNA。退火体系:Nuclease-Free Water 40 μL,Annealing Buffer for DNA Oligos(5×)20 μL,DNA oligo A(50 μmol/L)20 μL,DNA oligo B(50 μmol/L)20 μL,总体积100 μL。退火程序:95 ℃、2 min;95 ℃、1.5 min;-1 ℃/cycle;70 个循环;16 ℃保存。
1.3.7.2 Octet RED96e分子互作分析系统上机实验
将NTA传感器插入PBS中预湿10 min以上,第1步将传感器转入A Buffer(PBS)中进行第1次基线步骤(baseline 1),第2步将传感器转入用A Buffer稀释的PurL蛋白溶液(终质量浓度为160 μg/mL)中进行蛋白固化步骤(loading),第3步将传感器转入B Buffer(PBS+0.02% Tween+0.1% BSA)中进行第2次基线步骤(baseline 2),第4步将传感器转入用B Buffer稀释的DNA溶液中进行结合步骤(association),第5步将传感器转回B Buffer中进行解离步骤(dissociation)。各步骤的时间根据基线平衡情况以及固化情况进行调整。PurR蛋白实验时将第1步与第2步中的A Buffer换为C Buffer(20 mmol/L Tris-HCl、0.5 mol/L NaCl),蛋白固化步骤中PurR蛋白溶液终质量浓度为25 μg/mL。实验设置DNA浓度梯度为86.2、43.1、21.5、10.75、5.37 nmol/L。
1.3.8 EMSA实验
参考文献[30]并进行部分调整。通过PCR扩增启动子序列,纯化回收后测定核酸浓度。蛋白和DNA结合反应体系中,5×EMSA结合缓冲液3 μL,DNA片段200~250 ng,不同浓度梯度纯化后的蛋白,加入ddH2O补足至15 μL。室温放置30 min后,加入2 μL EMSA/Gel-Shift上样缓冲液进行非变性蛋白电泳,结束后GelRed染色并用凝胶成像仪观察。
2 结果与分析
2.1 重组克隆载体及重组表达载体的构建与鉴定

图 1 重组克隆质粒以及重组表达质粒的酶切鉴定Fig. 1 Identification of recombinant cloned plasmids and recombinant expression plasmids by enzymatic digestion
针对构建的克隆载体pMD 19-T simple-purR和pMD 19-T simple-purL以及表达载体pQE-30-purR和pQE-30-purL,用相应的酶分别进行分步酶切鉴定,电泳结果显示有两条明亮条带(图1)。用pMD 19-T simple通用引物以及pQE-30测序引物进行测序,测序结果用BLAST工具与全基因组测序结果进行比对,序列完全一致,表明克隆载体pMD 19-T simple-purR和pMD 19-T simple-purL,以及表达载体pQE-30-purR和pQE-30-purL已成功构建。
2.2 PurR蛋白和PurL蛋白的诱导表达及鉴定
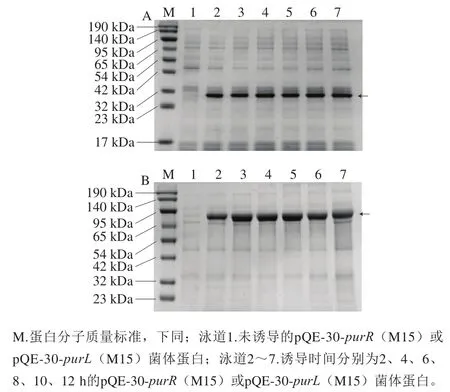
图 2 PurR蛋白(A)和PurL蛋白(B)的SDS-PAGEFig. 2 SDS-PAGE analysis of expressed recombinant PruR (A) and PurL (B)
如图2所示,与未诱导的菌体蛋白相比(泳道1),加入IPTG诱导剂后的菌体蛋白(2~7泳道)在箭头处出现明显的蛋白条带,与软件预测大小相近,PurR蛋白和PurL蛋白诱导表达成功。并且随诱导时间的延长,重组蛋白的表达量增加,诱导6 h的PurR蛋白和PurL蛋白表达量较高,6 h后蛋白表达量增加不明显,所以确定PurR蛋白和PurL蛋白的诱导时间为6 h。
2.3 PurR蛋白和PurL蛋白的纯化与定量
20 mmol/L以及50 mmol/L咪唑洗脱液都不会洗脱目的蛋白,并可以洗脱少量杂蛋白(图3)。对于PurR蛋白,200 mmol/L咪唑洗脱液就可以得到较高浓度的目的蛋白(图3A);对于PurL蛋白,咪唑浓度需达到300 mmol/L时才可得到较高浓度的蛋白(图3B),经此方法纯化的目的蛋白条带较单一,纯度较高。Bradford蛋白质量浓度测定试剂盒检测纯化后PurR蛋白质量浓度为0.74 mg/mL,PurL蛋白质量浓度为3.8 mg/mL,这是由于PurR蛋白是对破壁后的上清液进行纯化,但部分PurR蛋白以包涵体形式存在,纯化过程中会有部分损失,且在浓缩过程中浓度增加会有析出。
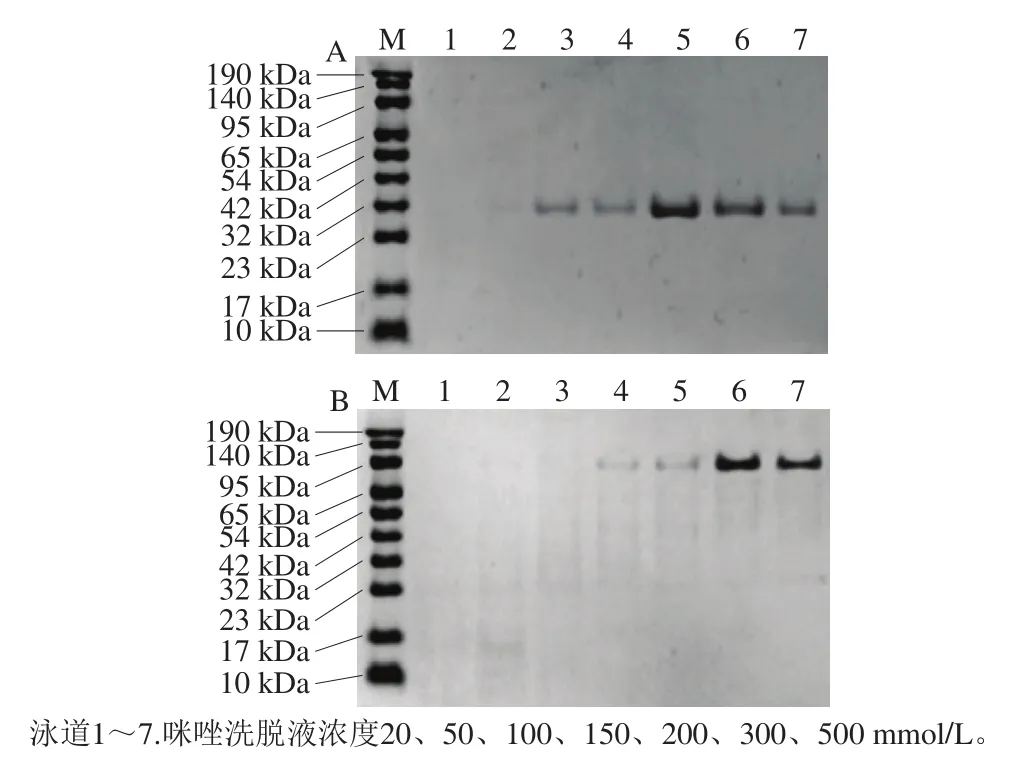
图 3 PurR蛋白(A)和PurL蛋白(B)经不同咪唑浓度洗脱液洗脱的目的蛋白SDS-PAGEFig. 3 SDS-PAGE analysis of purified PruR (A) and PurL (B) with different concentrations of imidazole as eluent
2.4 BLI实验结果

图 4 启动子与重组蛋白的结合实验结果Fig. 4 BLI results for binding between promoters and recombinant proteins
单浓度预实验结果表明,plnE启动子与PurR蛋白和PurL蛋白可能存在结合作用,plnG启动子与PurR蛋白可能存在结合作用,plnG启动子与PurL不存在结合作用。所以对3 组可能存在相互作用的启动子与表达蛋白进一步进行浓度梯度实验,根据预实验结果调整固化物PurR蛋白质量浓度为25 μg/mL,PurL蛋白质量浓度为160 μg/mL,分析物DNA浓度梯度为86.2、43.1、21.5、10.75、5.37 nmol/L。在association步骤以及dissociation步骤曲线平直,且DNA浓度不同时,结合与解离曲线基本重合,3 组浓度梯度实验均没有浓度依赖的结合解离过程(图4),没有观察到PurR和PurL蛋白与plnE启动子和plnG启动子的直接相互作用。
2.5 EMSA实验结果
EMSA检测PurR、PurL蛋白与plnE、plnG启动子的结合作用发现,与不加蛋白的对照组(泳道1)相比,无论何种蛋白质量浓度,DNA条带均没有出现滞后现象,没有出现蛋白与启动子片段结合的条带(图5)。结合BLI实验结果,发现通过原核表达得到的PurR和PurL蛋白在体外没有观察到与植物乳杆菌KLDS1.0391细菌素合成调控区域的plnE和plnG启动子直接的相互作用。

图 5 重组蛋白与启动子结合的EMSA实验结果Fig. 5 EMSA results for recombinant proteins binding to promoters
3 讨 论
研究表明,PlnC和PlnD是一对反应调节器,能够调控植物乳杆菌细菌素的合成,磷酸化的PlnC和PlnD能够结合到调控细菌素合成基因簇的启动子上,激活或抑制细菌素合成相关基因的表达[31]。对植物乳杆菌KLDS1.0391盐胁迫的初步研究发现,低浓度盐能够促进细菌素合成,但作用机制尚不明确,已有的细菌素调控机制无法解释这种现象,在这一过程中是否存在其他相关调控蛋白与细菌素合成调控区域结合进而控制细菌素的合成尚不清楚。研究调控蛋白与核酸的结合作用,能够更深入地理解基因表达调控过程,从分子层面进一步解释微生物的各种生理活动。目前研究调控蛋白与核酸相互作用的实验方法主要有体外与体内研究方法。徐会永[32]通过EMSA实验证明通过原核表达的LeClp蛋白与pks/nrps基因的启动子在体外具有结合作用;张良林等[26]通过BLI技术,在体外研究了原核表达的HetR蛋白与其靶DNA(PhetP与PhetZ)的结合情况及其结合亲和力。体外实验能够直观地显示蛋白与核酸是否具有结合作用,本实验目的是通过体外研究方法进行初步探究。在作用蛋白的确定上,本实验基于课题组前期的转录组测序分析,在低浓度盐胁迫条件下,purR与purL基因表达量显著下调,并且已有研究也表明purR基因在微生物的许多生理活动中起调控作用,具有全局调控机制;purL基因与微生物的生理活动也有着密切关系,解淀粉芽孢杆菌purL基因突变株的细菌素合成量显著升高,推测其对细菌素合成具有调控作用。基于以上原因,本研究先通过体外实验研究purR与purL基因对植物乳杆菌细菌素合成的调控作用。
通过基因重组技术,体外成功合成了PurR和PurL蛋白,为其体外与启动子的结合作用实验提供支撑。在BLI实验时,通常选择对DNA进行biotin修饰,将生物素化的DNA固化在SA传感器上,将蛋白作为分析物,但在实验过程中发现带有His标签的PurR和PurL蛋白与SA传感器具有非特异性结合,通过加入BSA与Tween 20无法消除存在的非特异性结合,所以不能选择SA传感器进行实验。Ni-NTA传感器能够捕获带有His标签的分子,所以尝试将带His标签的蛋白固化在Ni-NTA传感器上,将DNA作为分析物进行实验,结果没有观察到PurR蛋白和PurL蛋白与植物乳杆菌KLDS1.0391细菌素合成启动子直接的结合作用。BLI技术通过检测生物传感器底部膜厚度的变化,计算分子间的相互作用强度,将DNA作为分析物,结合在传感器底部,并不能引起较大膜层厚度的改变,因此还需要通过其他实验进一步验证。进一步EMSA实验,PurR和PurL蛋白与植物乳杆菌KLDS1.0391细菌素合成启动子也无直接的结合作用,所以体外实验表明,purR和purL基因可能并不直接参与细菌素合成的调控。有研究表明,乳酸乳球菌中的purR与枯草芽孢杆菌中的purR同源,后者是转录抑制蛋白,前者是转录激活蛋白[5],在植物乳杆菌KLDS1.0391中,推测purR也起转录激活作用,purR表达量下调引起purL等基因的表达下调,purL基因的表达量降低会影响焦磷酸硫胺素(thiamine pyrophosphate,TPP)与肌苷一磷酸(inosine monophosphate,IMP)的合成,Liu Quanli等[15]研究表明,解淀粉芽孢杆菌中purL基因突变株的TPP与IMP合成均受到抑制,菌株抑菌能力增加,向底物中添加TPP后菌株抑菌能力恢复到与野生菌株相同水平。因此,在2% NaCl胁迫条件下,植物乳杆菌KLDS1.0391中purR和purL表达量降低,推测有可能对细菌素合成量升高起到间接的调控作用。但是蛋白调控过程是一个涉及到菌体内多种因素的复杂的网络调控过程,菌体内环境复杂,菌体外研究所得到的结果不一定能真实反映菌体内的结合情况,还需进一步研究菌体内两种蛋白是否具有结合能力以及两种蛋白是否参与细菌素合成的调控。
4 结 论
体外成功构建重组蛋白PurR和PurL,EMSA实验以及BLI技术结果表明,2 个重组蛋白与植物乳杆菌KLDS1.0391细菌素合成基因的启动子在菌体外无直接结合作用,在菌体内的情况以及2 种蛋白对细菌素合成的调控是否属于间接调控作用,需进一步研究。
