球姜酮对照品的制备
2021-03-25吴相欢田民义赵晓歌陆廷亚邓国栋
吴相欢,田民义*,赵晓歌,陆廷亚,韦 凤,邓国栋
(1.贵州大学酿酒与食品工程学院,贵州 贵阳 550025;2.贵州省药食同源植物资源开发工程技术研究中心,贵州 贵阳 550025)
红球姜Zingiber zerumbet(L.) Smith 为姜科姜属植物,根茎具有芳香气味,主要分布在国内云南、广西、广东及国外斯里兰卡、尼泊尔、孟加拉、马来西亚、印度等地[1-2],既可药用,又可食用,其根茎能祛风解毒,治疗感冒、发烧、腹痛、胃痉挛、胃肠胀气、咳嗽、食欲不振、细菌感染、过敏、皮肤病等,并能提取芳香油作调合香精原料,嫩茎叶可当蔬菜[2-4]。药理研究表明,它具有抗肿瘤[5]、镇痛[6]、抗炎[7]、抗疟疾[8]、抗氧 化[9]、抗 微生物[10]等作用,球姜酮为其主要活性成分,属于倍半萜类,有着抗菌[5]、抗肿瘤[11]、抗炎[12]、抗高胆固醇[13]、抗乙酰胆碱酯酶活性[14],以及镇痛[15]、胃保护[16]、肝 保护[17]、改善糖尿病作用[18]。
目前,红球姜已被开发为香料和茶饮料产品,其挥发油开发为化妆品,并作为传统民间药被广泛使用,球姜酮为其主要药理活性成分,而且含量高,故可将其作为药材质量的评价指标。本实验旨在开发一种简单、便捷、高纯度的球姜酮制备方法,以期用于红球姜质量评价,并为该成分药理研究提供物质基础。
1 材料
1.1 试剂与药物 红球姜于2018 年10 月采自广西玉林,经贵州大学熊源新教授鉴定为姜科姜属红球姜Zingiber zerumbet(L.) Smith,标本保存于贵州省药食同源植物资源开发工程技术研究中心。薄层色谱硅胶(青岛海洋化工有限公司)。乙腈、甲醇为色谱纯(德国Merck 公司);其他试剂均为分析纯。
1.2 仪器 Agilent 1260 分析型高效液相色谱仪(美国Agilent Technologies 公 司);HP6890/5975C GC/MS 联用仪(美国安捷伦公司);JEOL ECX-400 核磁共振仪(日本电子株式会社);HP5973 质谱仪(美国惠普公司);FA2004型电子天平(上海良平仪器仪表有限公司)。
2 方法与结果
2.1 挥发油提取 称取500 g 鲜红球姜地下根茎,粉碎后置于2 L 圆底烧瓶中,加入1 L 蒸馏水,连接挥发油提取器,微沸4 h 后将提取器中的液体放入分液漏斗中,加入适量正己烷萃取,正己烷层用无水Na2SO4除水,滤过,减压浓缩除去溶剂,即得(0.51 g)。
2.2 挥发油GC-MS 鉴定 分析条件为HP6890/5975C GC/MS 联用仪,FB-5MSI 弹性石英毛细管柱(30 m×0.25 mm×0.25 μm);进样量1 μL;初始温度46 ℃,保留2 min,3 ℃/min升至190 ℃,以15 ℃/min 升至310 ℃,运行时间58 min;汽化室温度250 ℃;载气高纯He (99.999%),柱前压7.65 psi (1 psi=0.133 kPa);载气体积流量1.0 mL/min;分流比20 ∶1;溶剂延迟时间4.0 min;离子源EI 源,温度230 ℃;四极杆温度150 ℃;电子能量70 eV;发射电流34.6 μA;倍增器电压1 565 V;接口温度280 ℃;质量范围m/z29~500。通过质谱计算机数据系统检索、Nist2014 和Wiley275 标准质谱图比对,鉴定总离子流图(图1) 中各色谱峰对应的化学成分,峰面积归一化法测定球姜酮质量分数为77.8%。
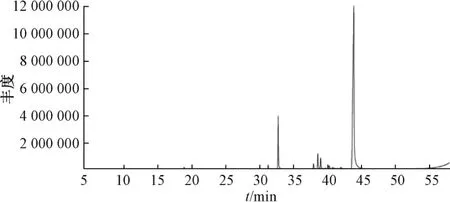
图1 挥发油GC-MS 色谱图
2.3 球姜酮纯化 挥发油中加入0.3 mL 正己烷,40 ℃水浴加热使其完全溶解,置于-22 ℃冰箱中冷藏15 min,析出晶体,减压抽滤,得白色粗品,加入0.2 mL 正己烷,40 ℃水浴加热溶解,置于-22 ℃冰箱中冷藏10 min,减压抽滤,得到0.32 g 纯品,纯度大于99%。
2.4 球姜酮结构鉴定 EI-MSm/z(%):218 [M+],203,189,175,163,150,135 (100),121,107,96,79,67,53,41。1H-NMR (CDCl3,400 MHz)δ:5.96~5.92 (1H,m,H-6),5.89 (1H,d,J=16.4 Hz,H-9),5.78 (1H,d,J=16.4 Hz,H-10),5.18 (1H,bd,J=15.1 Hz,H-2),2.43~2.30 (1H,m,H-5),2.30~2.12(4H,m,H-1,4,5),1.88~1.75 (1H,m,H-1),1.72(3H,s,H-13),1.46 (3H,s,H-12),1.13 (3H,s,H-14),0.99 (3H,s,H-15);13C-NMR (CDCl3,100 MHz)δ:42.4 (C-1),125.0 (C-2),136.3 (C-3),39.5 (C-4),24.5 (C-5),148.9 (C-6),137.9 (C-7),204.3 (C-8),127.2 (C-9),160.7 (C-10),37.9 (C-11),15.3 (C-12),11.8 (C-13),29.5 (C-14),24.3 (C-15),以上数据与文献[19] 报道一致,故鉴定为球姜酮。
2.5 球姜酮纯度检测
2.5.1 TLC 法 取球姜酮适量,甲醇制成1 mg/mL 供试品溶液,吸取2、5、10、15、20 μL 点于GF254硅胶薄层板上,以氯仿-甲醇(2 ∶1)、丙酮-甲醇(6 ∶1)、乙酸乙酯-丙酮-甲醇(4 ∶2 ∶1) 为展开剂在254 nm 波长下观察,碘蒸气显色。结果,在3 种展开剂、2 种显色条件下均呈现单一球姜酮斑点,未见杂质。
2.5.2 HPLC 法 分析条件为Agilent 1260 高效液相色谱仪,岛津Wonda Cract ODS-2 色谱柱(4.6×250 mm,5 μm);检测波长280 nm;流动相乙腈-水(80 ∶20);进样量10 μL;柱温25 ℃;体积流量1 mL/min。设定240、260、280、290、310 nm 5 个信号通道,用于考察球姜酮纯度。
称取10 mg 球姜酮,乙腈制成1 mg/mL 供试品溶液,取其和溶剂(乙腈) 各10 μL 进行测定,扣除溶剂干扰峰后采用归一化法计算球姜酮含量,结果见表1,可知在各波长下均大于99%。
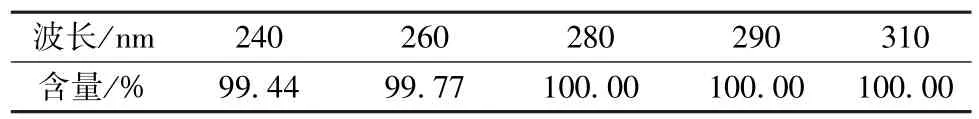
表1 球姜酮含量测定结果
2.6 稳定性试验 按照2015 年版《中国药典》 四部通则“原料药物与制剂稳定性试验指导原则”,分别对球姜酮进行室温、高湿、强光照试验,再按“2.2.2” 项下方法检测其纯度。
2.6.1 室温 取适量球姜酮至西林瓶中,敞口放置10 d,由于该成分具有挥发性,故将温度设定为室温(25 ℃),于第0、5、10 天取样检测,结果见表2。由此可知,球姜酮在该条件下放置10 d 后外观性状无明显变化,含量稳定。
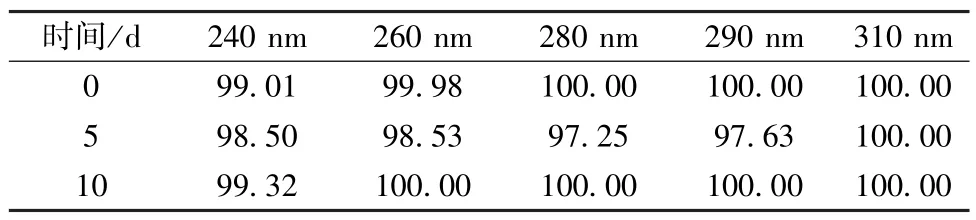
表2 室温试验结果(%)
2.6.2 高湿 取适量球姜酮至恒温恒湿培养箱中,在温度25 ℃、相对湿度90%下放置10 d,于第0、5、10 天取样检测,结果见表3。由此可知,球姜酮在该条件下放置10 d后外观性状无明显变化,含量稳定,吸湿增重小于5%。
2.6.3 强光照 取适量球姜酮至光照箱中,在4 500 lx 下放置10 d,于第0、5、10 天取样检测,结果见表4。由此可知,球姜酮在该条件下放置10 d 后外观性状无明显变化,含量稳定。
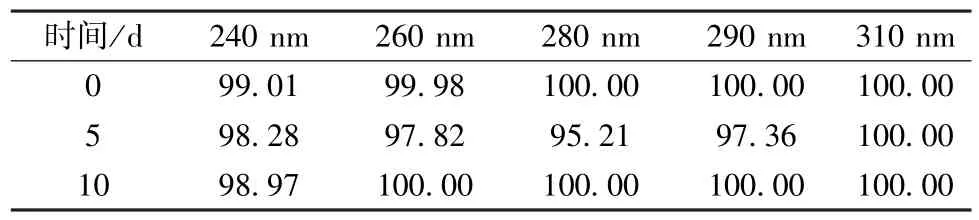
表3 高湿试验结果(%)
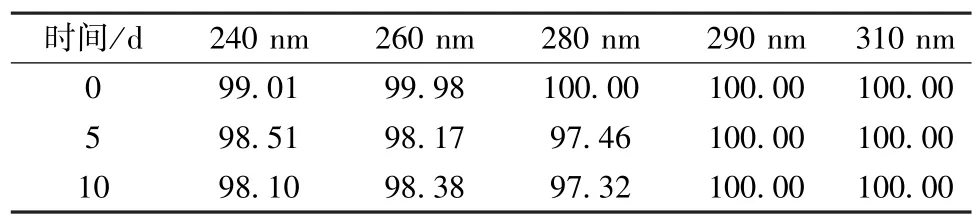
表4 强光照试验结果(%)
2.7 球姜酮含量测定
2.7.1 检测波长选择 本实验考察了240、260、280、290、310 nm 波长,发现供试品溶液在280 nm 处有最大吸收,故选择其作为检测波长。
2.7.2 流动相选择 取适量球姜酮供试品溶液,分别以乙腈-0.25%乙酸(70 ∶30)、甲醇-0.05%甲酸(80 ∶20)、乙腈-甲醇-水(60 ∶15 ∶25)、乙腈-水(80 ∶20)、甲醇-水(80 ∶20) 为流动相,在280 nm 波长处检测,结果见表5。由此可知,乙腈-水(80 ∶20) 为洗脱时对称性较好,故以其为流动相。

表5 流动相考察结果
2.7.3 柱温选择 取适量供试品溶液,在柱温25、35、48 ℃下进行考察,结果见表6。由此可知,当柱温为25 ℃时基线稳定,对称性较好。
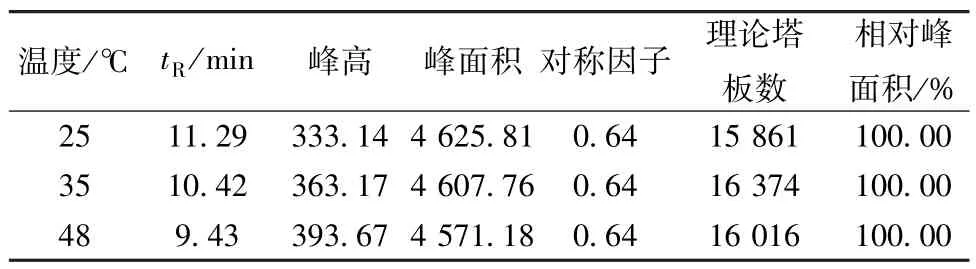
表6 柱温考察结果
2.7.4 体积流量选择 取适量供试品溶液,在体积流量0.8、1.0、1.2 mL/min 下进行考察,结果见表7。由此可知,当体积流量为1.0 mL/min 时对称性较好。
2.7.5 色谱柱选择 取适量供试品溶液,在检测波长280 nm、柱温25 ℃、流动相乙腈-水(80 ∶20)、体积流量1 mL/min、进样量10 μL 色谱条件 下,采 用Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)、Wonda Cract ODS-2 (4.6 mm × 250 mm,5 μm)、Eclipse XDB-C18(4.6 mm×250 mm,5 μm) 色谱柱进行考察,结果见表8。由此可知,Wonda Cract ODS-2 (4.6 mm×250 mm,5 μm)色谱柱对称性较好。
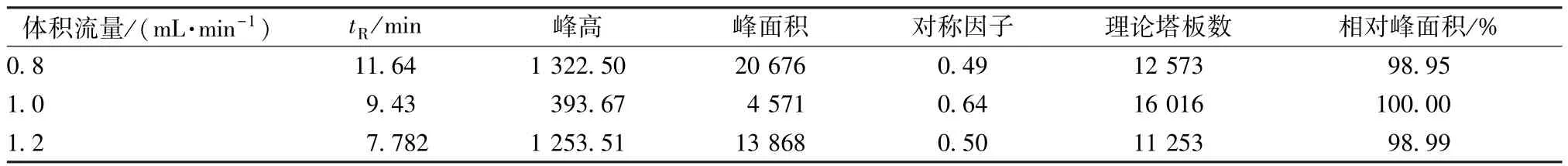
表7 体积流量考察结果
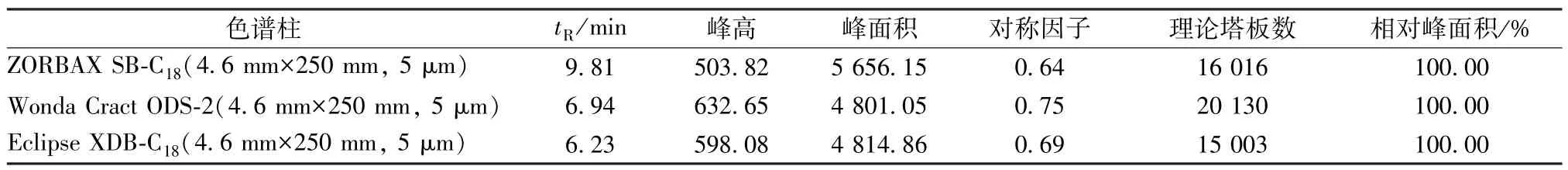
表8 色谱柱选择
3 结论
本实验所得球姜酮对照品纯度大于99%,符合国家标准,而且制备路线简单稳定,可用于含该成分药材及相关制剂的质量控制和药效物质基础研究。