有机肥施加对红壤中反硝化细菌nirK基因多样性影响①
2021-03-23张晨阳滕齐辉崔中利李顺鹏
张晨阳,滕齐辉,曹 滢,崔中利,李顺鹏,曹 慧
有机肥施加对红壤中反硝化细菌基因多样性影响①
张晨阳,滕齐辉,曹 滢,崔中利,李顺鹏,曹 慧*
(南京农业大学生命科学学院/农业农村部农业环境微生物工程重点实验室,南京 210095)
为了解有机肥施用对于红壤中反硝化细菌群落结构的影响,设置了4个处理:CK(不施肥)、LM(低量有机肥)、ML(高量有机肥+石灰)和HM(高量有机肥)进行研究,并通过末端限制性片段长度多态性(T-RFLP)和克隆文库的DNA测序估计了基因的多样性。结果表明:各处理分别挑选的288个阳性克隆子可分为78个类型,各处理文库中的优势类群属于同一种类型,在CK处理中的占比为51%,在其他3个处理中的占比依次为33%、32% 和27%。4个文库之间两两的相似性在37.50% ~ 45.34%,系统进化树分析表明,51个OTUs测序结果中6个OTUs与苍白杆菌属的相似性最高,占测序总数的11.8%;其余45个OTUs属于未培养类型,占测序总数的88.2%。有机肥添加有助于提高基因的多样性,并且出现了红壤原始环境中未出现的反硝化细菌类型。
反硝化细菌;基因;RFLP 分析;红壤;有机肥施加
红壤在我国热带和亚热带地区广泛分布,是我国最主要的土壤类型之一,在全国范围内共有148万km2,占耕地总面积的36%[1]。红壤由于风化和淋溶作用,使得土壤的酸性较强、养分匮乏,缺乏许多营养元素,尤其是氮、磷、钾、钙、镁、硫、锌和铜[2-3]。为了提高红壤的肥力,通常采用有机肥来改善其质量,添加到土壤中的有机肥可以改善土壤性质,例如土壤团聚体、持水能力、土壤容重和肥力等[4-6]。康国栋等[7]研究发现有机肥的施加可以促进红壤有机质活性组分的提高,对于土壤的培肥至关重要。石灰也常常被用来修复酸性土壤。在热带地区,施用少量的石灰就可以缓解铝毒性和/或钙缺乏状况,从而增加作物产量[8],石灰的施加对于红壤中钾的含量也有一定的影响[9]。由于红壤在干湿环境下的特殊性质,容易滋生反硝化微生物[10],而施肥被认为是土壤微生物演替的重要驱动因素[11],包括化肥和有机肥在内的氮肥施用,可以促进硝化作用与反硝化作用,导致氮素的流失[12-14]。因此研究有机肥添加对于红壤中反硝化细菌的影响有较大意义。
反硝化作用是氮循环中的关键组分,是在微氧或无氧环境中一种由微生物介导的逐步还原过程,其中硝酸盐(NO– 3)依次还原为亚硝酸盐(NO– 2),一氧化氮(NO),一氧化二氮(N2O),最后还原为氮气(N2)[15]。反硝化作用的4个生物过程对应着4种在反硝化过程中互相独立的关键酶,分别是硝酸还原酶、亚硝酸还原酶、氧化氮还原酶和氧化亚氮还原酶,对应的功能基因分别为、、和。而亚硝酸还原酶参与的反应是区分硝酸盐呼吸菌和反硝化细菌的标志性反应,同时这一步反应还会释放出臭氧被还原的催化剂气体,因此是反硝化过程中的重要限速步骤[16-18]。亚硝酸还原酶具有两种由不同基因(和)编码的结构形态,许多研究将这两种基因用于研究反硝化细菌群落。研究发现,含有基因的微生物以假单胞菌(sp)为主,而基因则存在于很多分布较远的微生物中[17];另外,也有研究发现基因对环境因子的变化相较于基因更为敏感,环境因子如NO– 3、全氮、含水量等的改变会影响含有基因的反硝化细菌的多样性[19-20]。因此,研究环境中的基因可以更为真实地了解环境中反硝化细菌群落结构的情况,也有助于了解环境因子对于反硝化细菌群落的综合作用。
有研究者发现,土壤中由于反硝化作用而损失的氮肥占其总量的20% ~ 30%[21]。氮肥是土壤氧化亚氮排放的主要来源,而这种气体的排放导致全球气候发生变化[22],通过反硝化和氨挥发损失的氮素量随氮肥用量的增加而增加[23]。在之前的研究中研究者对有机肥的施用方式与有机肥的类型对于反硝化细菌的影响进行了研究[12-14, 24],但是对于红壤中施用有机肥对于含有基因的反硝化细菌影响尚无定论。本研究利用限制性片段长度多态性聚合酶链反应(PCR-RFLP)技术研究长期(4年)施加有机肥和石灰的红壤中基因群落结构的改变,探讨红壤地区反硝化细菌的多样性及其重要影响因素,为红壤陆地生态系统研究提供部分参考。
1 材料与方法
1.1 试验设计及样品采集
采样地点位于江西省鹰潭市的中国科学院红壤生态实验站内(116°55′30″ E,28°15′20″ N),试验土壤为第四纪红黏土发育的红壤(黏化湿润富铁土),试验小区为长2 m×宽2 m的水泥池,自2002年起,每年种植一季玉米(山东登海一号)。试验使用养殖厂猪粪作为有机肥,设置4个处理:①CK,不施肥;②LM,低量有机肥,折合纯N 150 kg/(hm2·a);③ML,高量有机肥+石灰,折合纯N 600 kg/(hm2·a) + 石灰1 000 kg/(hm2·a) (考虑到石灰处理对于土壤的影响及作用时间,2002年和2005年各施加一次石灰);④HM,高量有机肥,折合纯N 600 kg/(hm2·a)。每处理3个重复。2006年7月下旬收获作物一周后进行土壤样品采集,每个小区土壤样品均为9点混合样品,用不锈钢土钻(直径2 cm) 钻取0 ~ 15 cm的土样,用四分法留取土样,低温冷藏带回实验室,去除石头和植物根系,取一部分混合土壤样品风干后过筛(<2 mm)用于理化性质测定;另取一部分保存于–70℃冰箱中用于土壤微生物总DNA提取。试验土壤理化性质参见文献[25]。
1.2 供试菌株与试剂
本试验除大肠杆菌DH10B、限制性内切酶HhaI和RsaI购自大连宝生物工程有限公司外,其余所有在试验中使用的菌株和试剂均与本实验室之前的研究中使用的一致[26]。
1.3 反硝化细菌nirK基因片段的扩增与克隆文库的构建
使用Zhou等[27]提出的直接提取法对土壤样品总DNA进行提取。采用引物(5'- GGMATGGTKCCSTGGCA -3')和(5'- GCCTCGATCAGRTTRTGG -3')用于直接扩增土壤总DNA中的基因片段[28]。PCR反应体系与汪峰等[26]研究中使用的体系一致,PCR扩增程序采用“降落”PCR 程序。反应条件:94℃预变性5 min;94℃变性30 s,55℃退火30 s,72℃延伸1 min,10 个循环,每个循环降0.5℃;后再接50℃退火30 s,72℃延伸1 min,10 个循环,每个循环升0.5℃; 52℃退火30 s,72℃延伸1 min,10 个循环,最后72℃延伸10 min。使用1% 的琼脂糖凝胶电泳检测PCR产物的条带大小和浓度,为了避免单次扩增带来的偏差并获得足够的PCR产物用于克隆,对每个DNA样品进行3次重复扩增,然后合并使用回收试剂盒回收纯化目的片段,并根据制造商的方案克隆到pMD18-T载体中。将质粒转化到感受态DH10B中,使用蓝白斑筛选,从4个处理中各挑选出288个阳性克隆子,建立基因片段的克隆文库,所有的克隆文库验证后转移至96孔细胞培养板,加等体积的30% 甘油混合后于–70℃保存。
1.4 限制性内切酶片段长度多态性分析
直接挑取阳性转化子菌体,使用菌体PCR的方式利用上述引物再次进行片断的扩增,分别用HhaI和RsaI两种限制性内切酶对PCR产物进行酶切(37 ℃,3 h)。使用8% 的聚丙烯酰胺凝胶电泳对酶切产物进行分离,经硝酸银染色、凝胶成像整理后即为反硝化基因指纹图谱。所得到的指纹图谱用上海山富科技服务有限公司的(Bio-science)GIS凝胶成像分析系统分析并辅以人工分析。以基因片段多态图像为基础进行聚类,将两种酶切产物带型一致的基因型分为一类,每一个基因型即为一个操作分类单位(operational taxonomic unit,OTU)。
通过统计分析方法对4个处理进行多样性比较,计算得到群落的文库覆盖率 (Coverage C)、香农指数、辛普森指数、均匀度和丰富度指数。
1.5 序列测定和系统发育树构建
对4个处理文库中的代表性酶切克隆子进行了测序,共挑取54个克隆子,测定其插入序列全长(515 bp左右)。测序工作由上海生工生物技术公司完成,测序引物为pMD18-T vector多克隆位点两端的通用引物(primer RV-M 和primer M13-4)。将测序获得的序列提交至NCBI(National Center for Biotechnology Information),得到登录号为EU790823 ~ EU790873,使用BLAST对GenBank(http://www.ncbi. nlm.nih.gov)已经存在的序列进行同源性比对。经过嵌合体检查后,将确定为反硝化基因的序列用BioEdit v7.0.1转换成FASTA 格式。同时,根据BLAST同源性比对的结果,从核酸数据库中下载同源性高的序列以及不同分类来源的代表性序列,然后按照汪峰等[26]研究中的方法进行系统进化树构建,从而进行系统发育分析。
2 结果与分析
2.1 nirK基因文库的酶切类型
对CK、LM、ML和HM 4个处理的土壤DNA进行PCR扩增,构建基因克隆文库,通过RFLP分析发现,这些基因具有丰富的物种多样性。根据获得的清晰RFLP图谱,各个处理分别挑选的288个克隆子可分为78种类型,并将这78种类型分别命名为CK-A至CK-N,LM-A至LM-S,ML-A至 ML-T和HM-A至HM-Y,4个克隆文库聚类比例见表1。CK文库中占比最大的类群即为其优势类群,占比达到了51%,超过了CK文库总数的一半,LM文库中优势类群所占比例为33%,ML文库中优势类群的比例为32%,HM文库中优势类群占总数的27%。优势类群是这4个文库中共有的,并且其相对含量随着有机肥的添加而逐渐降低;在4个文库中出现了部分特有的类型,从各处理类群数也可看出随着肥力的增加反硝化细菌的类群是逐渐增加的。
表2是4个基因文库之间的群落相似性矩阵,通过相似性比较可以看出不同的处理导致群落间变化的差异程度。4个文库两两之间的相似性在37.50% ~ 45.34%;随着有机肥添加量的增加相似性值下降。说明整体上4个处理依旧存在着很大的共性部分,但有机肥的添加可以改变反硝化细菌群落。
2.2 nirK基因文库的多样性指数
图1是4个基因文库的丰富度曲线,从图1中可以看出,4个文库的趋势线已经趋于平缓,说明库容量已经达到了比较饱和的程度。由表3可知,4个文库的文库覆盖率分别为88.23%(CK)、86.57% (LM)、87.84%(ML)和80.82%(HM),均大于80%,说明本研究所建立的基因文库可以较好地代表红壤环境中反硝化细菌的群落组成。
从4个文库中的多样性指数可以看出,红壤中存在着丰富的反硝化微生物,各文库之间多样性指数差异比较明显。CK处理的基因多样性指数最低,HM处理的最高,而LM和ML处理的多样性指数比较接近,反硝化微生物的多样性随肥力程度的提高而逐渐增加。从文库的均匀度和丰富度来看,与文库的多样性指数趋势一致,说明有机肥量的增加使得反硝化细菌变得更加丰富且使得各类反硝化微生物趋向于更均匀。香农指数与环境因子的相关性分析结果见表4,仅碳氮比与香农指数之间呈显著正相关,其他各环境因子与香农指数之间无显著相关关系。
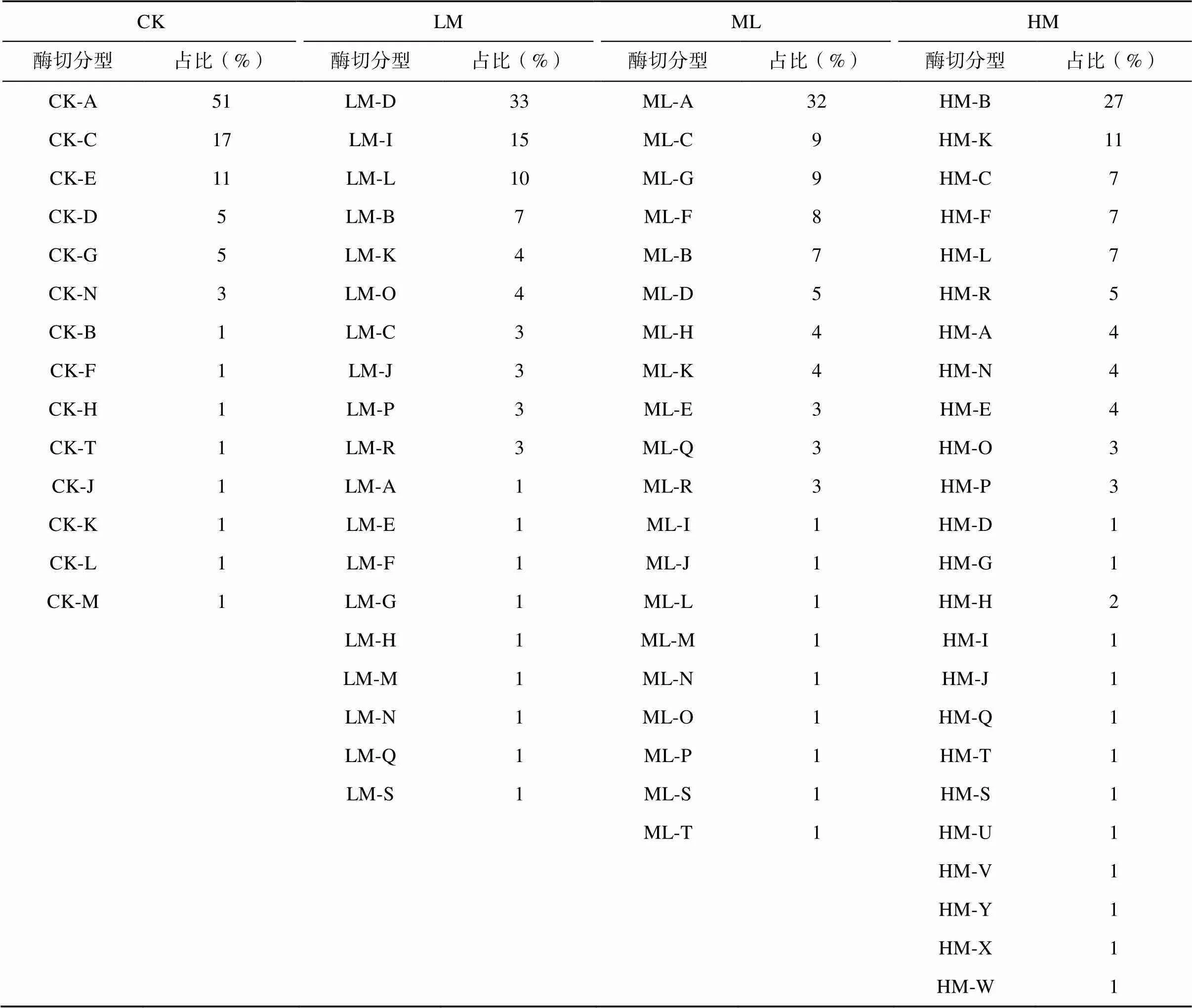
表1 4个nirK克隆文库酶切分型
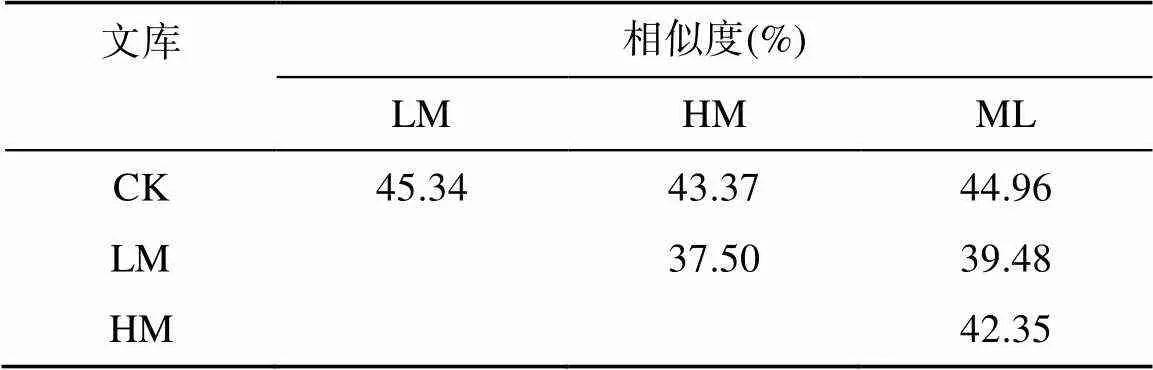
表2 4个nirK基因文库之间的群落相似性矩阵
2.3 系统进化树的构建与系统发育分析
本文将测序的54个OTUs通过NCBI的BLAST比对去除了3个假阳性结果,得到51个正确的基因序列,在Genbank上选择了部分与测定序列较为接近的序列,通过BioEdit 和Mega软件用这些序列构建系统进化树(图2),进行系统发育分析。结果表明,测序的基因彼此之间的相似性在60%~100%,与已知序列进行同源性比较,没有达到100% 的,说明这些序列可能是之前未被描述过的基因序列;这些OTUs主要被分为3个簇,大部分的OTUs出现在cluster 1中,占总克隆数的78.4%,HM处理在cluster1中占15%,其他3个处理各占25% ~ 30%,同时还发现在cluster 2和cluster 3中均未出现CK样品中的克隆子,其他3个处理各占30% 左右;红壤中反硝化细菌具有高度多样性,51个OTUs分属于苍白杆菌属(sp.)和未培养的细菌,并且绝大多数与未培养细菌的相似度更高。
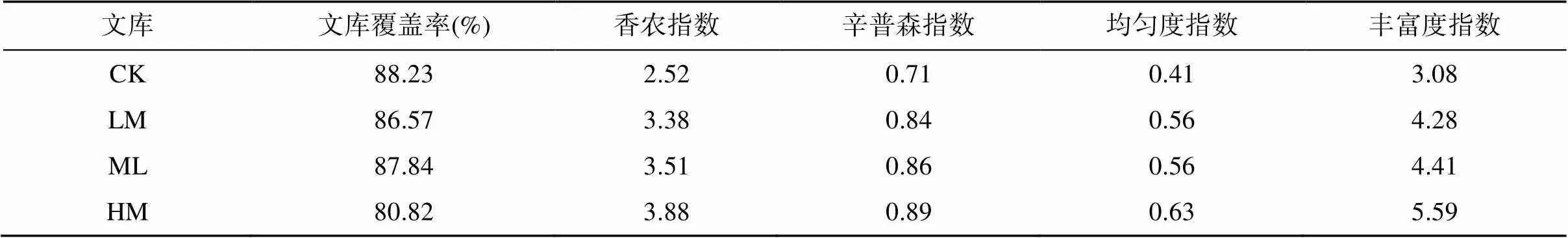
表3 4个nirK基因文库的群落结构多样性指数

表4 香农指数与环境变量的Pearson相关性
注:*表示在<0.05 水平(双侧)显著相关,=4。
3 讨论
反硝化过程是由微生物将环境中被固定的氮素重新释放到大气中,从而实现自然界生态系统的氮素循环过程,是自然界氮素循环的重要组成部分[29]。研究表明,反硝化作用产生了大量的温室气体,如NO和N2O[30],导致地球生物圈中氮素损失较大,自然环境的补给量远小于其损失程度,所以全球氮素预算处于极不平衡的状态[31-33]。红壤由于其养分匮乏、有机物质含量较低,因此向红壤中施肥是提高红壤经济效益的有效方式[2,5],但是关于有机肥添加对于红壤中含有基因的反硝化细菌群落影响的研究较少。本研究以基因作为分子标记,采用PCR-RFLP方法研究了多种有机肥添加处理对于红壤反硝化群落结构多样性的影响,结果表明,4个不同处理文库之间的群落结构组成有较大的差异,两两处理之间的相似性在37.50% ~ 45.34%。有研究认为当群落相似性大于60% 时属于相似性较好的群落[34],而本试验中相似值均小于60%,说明这4个基因文库之间的相似度均较低。在4个基因文库中优势类群所占比例依次为CK 51%、LM 33%、ML 32% 和HM 27%,而各文库中基因类群的数量依次为14种、19种、20种和25种。通过对香农指数和多样性指数等分析发现,各指数的变化情况相同,均为HM>ML>LM>CK,和图1中展示的结果相一致。Yang等[35]研究发现,氮含量较高的土壤有助于增加含有亚硝酸盐还原酶基因的反硝化菌的丰度;Chen等[36]研究发现,有机物质的添加可以增加反硝化菌群的丰度,并认为有机质的添加和碳含量的增加对于土壤中微生物的生存和繁衍具有积极影响。有机物质的添加可以提供丰富和平衡的营养物质(如高有机碳和一些无机盐),有利于氮循环中涉及的微生物的生长,本研究结果的趋势与之相同。
反硝化细菌群落香农指数与环境因子的Pearson相关性分析结果显示,仅碳氮比与香农指数呈显著正相关,其他环境因子与香农指数相关性不显著,与Yang等[37]的研究结果不符。而邓玉峰等[38]研究发现石灰的施加会降低细菌的数量,这可能导致了ML处理中反硝化细菌类群数量的减少,但ML处理各环境因子的值仍然较高,因此可能导致相关性检验不显著。
从本研究系统发育树可以看出,不同的有机肥添加处理土壤中反硝化细菌群落结构主要由-变形菌纲的苍白杆菌属(sp)和未培养的细菌组成,其中与未培养细菌相关的片段占到总数的88.2% 以上,与李刚等[39]的研究结果一致,但本研究中未培养细菌相关的比例更高,说明红壤中出现了丰富的反硝化细菌,这可能与红壤的性质有关系。红壤地区降水量丰富,雨季土壤常处于淹水的环境,干旱季节土壤又极易板结、不透气,时常处于厌氧的环境,有利于滋生反硝化微生物,导致反硝化作用的进行,反硝化基因也表现出丰富的程度[10]。大量研究发现,不同环境下反硝化细菌许多都属于未培养细菌。Henry等[40]对56个基因序列系统分析,发现多数基因序列与目前已知的可培养反硝化微生物的基因序列不同,Yoshida等[41]研究水稻土中含有基因的反硝化细菌群落,发现大部分细菌都与未培养细菌相关。本研究系统发育树分析将同属于苍白杆菌属的两个种聚成两类,这可能是因为这两个种中基因的分类较远。有研究者对和16S rRNA基因的系统发育分析结果表明,基因与16S rRNA基因不一致[42-43]。与苍白杆菌属聚类较近的多为CK处理中的克隆子,然后依次为LM、ML和HM处理,这说明不同的施肥条件改变了反硝化细菌的组成,但是具体的相关关系仍需要更为先进的分子生物学技术进行研究。
4 结论
通过向红壤中添加有机肥可以增加含基因的反硝化细菌多样性,并且反硝化细菌的类群是随着有机肥的添加量增加而增加,但是向添加有机肥的处理中添加石灰可以降低反硝化细菌类群数量;相似性分析和系统发育树表明,有机肥的添加改变了红壤中反硝化细菌的群落组成,但是并未改变反硝化细菌的优势菌群类型,未培养细菌是红壤中反硝化细菌的主要组成部分。
[1] 黄国勤, 赵其国. 红壤生态学[J]. 生态学报, 2014, 34(18): 5173–5181.
[2] Liu J, Liu M, Wu M, et al. Soil pH rather than nutrients drive changes in microbial community following long-term fertilization in acidic Ultisols of Southern China[J]. Journal of Soils and Sediments, 2018, 18(5): 1853–1864.
[3] Baligar V C, Fageria N K, Eswaran H, et al. Nature and properties of red soils of the world[M]//The Red Soils of China. Dordrecht: Springer Netherlands, 2004: 7–27.
[4] 柳开楼, 黄晶, 张会民, 等. 长期施肥对红壤旱地团聚体特性及不同组分钾素分配的影响[J]. 土壤学报, 2018, 55(2): 443–454.
[5] Zebarth B J, Neilsen G H, Hogue E, et al. Influence of organic waste amendments on selected soil physical and chemical properties[J]. Canadian Journal of Soil Science, 1999, 79(3): 501–504.
[6] Franzluebbers A J. Water infiltration and soil structure related to organic matter and its stratification with depth[J]. Soil and Tillage Research, 2002, 66(2): 197–205.
[7] 康国栋, 魏家星, 邬梦成, 等. 有机物料施用对旱地红壤作物产量和有机质活性组分的影响[J]. 土壤, 2017, 49(6): 1084–1091.
[8] Celik I, Ortas I, Kilic S. Effects of compost, mycorrhiza, manure and fertilizer on some physical properties of a Chromoxerert soil[J]. Soil and Tillage Research, 2004, 78(1):59–67.
[9] 韩天富, 王伯仁, 张会民, 等. 长期施肥及石灰后效对不同生育期玉米根际钾素的影响[J]. 土壤学报, 2017, 54(6): 1497–1507.
[10] 滕齐辉. 有机肥施用对红壤氮素循环微生物相关基因多样性的影响[D]. 南京: 南京农业大学, 2008.
[11] Cruz A F, Hamel C, Hanson K, et al. Thirty-seven years of soil nitrogen and phosphorus fertility management shapes the structure and function of the soil microbial community in a Brown Chernozem[J]. Plant and Soil, 2009, 315(1/2): 173–184.
[12] Dambreville C, Hallet S, Nguyen C, et al. Structure and activity of the denitrifying community in a maize- cropped field fertilized with composted pig manure or ammonium nitrate[J]. FEMS Microbiology Ecology, 2006, 56(1): 119–131.
[13] Enwall K, Philippot L, Hallin S. Activity and composition of the denitrifying bacterial community respond differently to long-term fertilization[J]. Applied and Environmental Microbiology, 2005, 71(12): 8335– 8343.
[14] Wolsing M, Priemé A. Observation of high seasonal variation in community structure of denitrifying bacteria in arable soil receiving artificial fertilizer and cattle manure by determining T-RFLP of nir gene fragments[J]. FEMS Microbiology Ecology, 2004, 48(2): 261–271.
[15] Zumft W G. Cell biology and molecular basis of denitrification[J]. Microbiology and Molecular Biology Reviews: MMBR, 1997, 61(4): 533–616.
[16] 康鹏亮, 张海涵, 黄廷林, 等. 湖库沉积物好氧反硝化菌群脱氮特性及种群结构[J]. 环境科学, 2018, 39(5): 2431–2437.
[17] Braker G, Zhou J Z, Wu L Y, et al. Nitrite reductase genes (and) as functional markers to investigate diversity of denitrifying bacteria in Pacific northwest marine sediment communities[J]. Applied and Environmental Microbiology, 2000, 66(5): 2096– 2104.
[18] Sun Y L, Li A, Zhang X N, et al. Regulation of dissolved oxygen from accumulated nitrite during the heterotrophic nitrification and aerobic denitrification of Pseudomonas stutzeri T13[J]. Applied Microbiology and Biotechnology, 2015, 99(7): 3243–3248.
[19] Smith J M, Ogram A. Genetic and functional variation in denitrifier populations along a short-term restoration chronosequence[J]. Applied and Environmental Microbiology, 2008, 74(18): 5615–5620.
[20] Han B, Ye X H, Li W, et al. The effects of different irrigation regimes on nitrous oxide emissions and influencing factors in greenhouse tomato fields[J]. Journal of Soils and Sediments, 2017, 17(10): 2457– 2468.
[21] Fillery I R P. Biological denitrification[M]//Gaseous Loss of Nitrogen from Plant-Soil Systems. Dordrecht: Springer Netherlands, 1983: 33–64.
[22] Hofstra N, Bouwman A F. Denitrification in agricultural soils: summarizing published data and estimating global annual rates[J]. Nutrient Cycling in Agroecosystems, 2005, 72(3): 267–278.
[23] 王书伟, 颜晓元, 单军, 等. 利用膜进样质谱法测定不同氮肥用量下反硝化氮素损失[J]. 土壤, 2018, 50(4): 664–673.
[24] Chen Z, Luo X Q, Hu R G, et al. Impact of long-term fertilization on the composition of denitrifier communities based on nitrite reductase analyses in a paddy soil[J]. Microbial Ecology, 2010, 60(4): 850–861.
[25] Teng Q H, Sun B, Fu X R, et al. Analysis ofgene diversity in red soil amended with manure in Jiangxi, South China[J]. Journal of Microbiology (Seoul, Korea), 2009, 47(2): 135–141.
[26] 汪峰, 曲浩丽, 丁玉芳, 等. 三种农田土壤中氨氧化细菌基因多样性比较分析[J]. 土壤学报, 2012, 49(2): 347–353.
[27] Zhou J, Bruns M A, Tiedje J M. DNA recovery from soils of diverse composition[J]. Applied and Environmental Microbiology, 1996, 62(2): 316–322.
[28] Braker G, Fesefeldt A, Witzel K P. Development of PCR primer systems for amplification of nitrite reductase genes (and) to detect denitrifying bacteria in environmental samples[J]. Applied and Environmental Microbiology, 1998, 64(10): 3769–3775.
[29] 孙建光, 高俊莲, 马晓彤, 等. 反硝化微生物分子生态学技术及相关研究进展[J]. 中国土壤与肥料, 2007(2): 7–12.
[30] Shoun H, Tanimoto T. Denitrification by the fungus Fusarium oxysporum and involvement of cytochrome P-450 in the respiratory nitrite reduction[J]. The Journal of Biological Chemistry, 1991, 266(17): 11078–11082.
[31] Ganeshram R S, Pedersen T F, Calvert S E, et al. Large changes in oceanic nutrient inventories from glacial to interglacial periods[J]. Nature, 1995, 376(6543): 755.
[32] Altabet M A, Curry W B. Testing models of past ocean chemistry using foraminifera15N/14N[J]. Global Biogeochemical Cycles, 1989, 3(2): 107–119.
[33] Middelburg J J, Soetaert K, Herman P M J, et al. Denitrification in marine sediments: a model study[J]. Global Biogeochemical Cycles, 1996, 10(4): 661–673.
[34] 王奇赞, 徐秋芳, 姜培坤, 等. 天目山毛竹入侵阔叶林后土壤细菌群落16S rDNA V3区片段PCR的DGGE分析[J]. 土壤学报, 2009, 46(4): 662–669.
[35] Yang C, Hamel C, Gan Y T. Incongruous variation of denitrifying bacterial communities as soil N level rises in Canadian canola fields[J]. Applied Soil Ecology, 2015, 89: 93–101.
[36] Chen Z, Hou H J, Zheng Y, et al. Influence of fertilisation regimes on a nosZ-containing denitrifying community in a rice paddy soil[J]. Journal of the Science of Food and Agriculture, 2012, 92(5): 1064– 1072.
[37] Yang Y D, Zhao J, Jiang Y, et al. Response of bacteria harboring nirS and nirK genes to different N fertilization rates in an alkaline northern Chinese soil[J]. European Journal of Soil Biology, 2017, 82: 1–9.
[38] 邓玉峰, 田善义, 成艳红, 等. 模拟氮沉降下施石灰对休耕红壤优势植物根际土壤微生物群落的影响[J]. 土壤学报, 2019, 56(6): 1449–1458.
[39] 李刚, 修伟明, 王杰, 等. 不同植被恢复模式下呼伦贝尔沙地土壤反硝化细菌基因组成结构和多样性研究[J]. 草业学报, 2015, 24(1): 115–123.
[40] Henry S, Baudoin E, López-Gutiérrez J C, et al. Quantification of denitrifying bacteria in soils bygene targeted real-time PCR[J]. Journal of Microbiological Methods, 2004, 59(3): 327–335.
[41] Yoshida M, Ishii S, Otsuka S, et al. Temporal shifts in diversity and quantity ofandin a rice paddy field soil[J]. Soil Biology and Biochemistry, 2009, 41(10): 2044–2051.
[42] Jones C M, Stres B, Rosenquist M, et al. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification[J]. Molecular Biology and Evolution, 2008, 25(9): 1955–1966.
[43] Heylen K, Gevers D, Vanparys B, et al. The incidence ofandand their genetic heterogeneity in cultivated denitrifiers[J]. Environmental Microbiology, 2006, 8(11): 2012–2021.
Effect of Organic Manure Application on Diversity ofGene in Denitrifying Bacteria in Red Soil
ZHANG Chenyang, TENG Qihui, CAO Ying, CUI Zhongli, LI Shunpeng, CAO Hui*
(Key Laboratory of Agricultural Environmental Microbiology, Ministry of Agriculture, College of Life Science, Nanjing Agricultural University, Nanjing 210095, China)
In order to understand the effect of organic manure application on the structure of denitrifying bacteria in red soil, four treatments were designed and compared: 1) CK, no manure; 2) LM, low organic manure; 3) ML, high organic manure + lime; and 4) HM, high amount organic manure. The diversity of thegene was estimated by terminal restriction fragment length polymorphism (T-RFLP) and DNA sequencing of the cloned library. The results showed thatthe 288 clones selected in each treatment can be divided into 78 clusters, and the dominant groups in eachlibrary were of the same cluster, accounting for 51% of the CK, while the other three treatments were 33%, 32% and 27%, respectively. The similarity between the four libraries was between 37.50% and 45.34%. The phylogenetic tree analysis of the 51 OTUs showed that 6 OTUs had the highest similarity withsp., accounting for 11.8% of the total number of sequencing. The remaining 45 OTUs belonged to the uncultured cluster, accounting for 88.2% of the total number of sequencing. Organic manure addition increased the diversity of thegene, but some types of denitrifying bacteria didn’t appear in the original red soil environment.
Denitrifying bacteria;gene; RFLP; Red soil; Application of organic manure
Q938.1
A
10.13758/j.cnki.tr.2021.01.010
张晨阳, 滕齐辉, 曹滢, 等. 有机肥施加对红壤中反硝化细菌基因多样性影响. 土壤, 2021, 53(1): 72–79.
国家自然科学基金项目(41371262;40871125)资助。
(hcao@njau.edu.cn)
张晨阳(1994—),男,山西长治人,硕士研究生,研究方向为土壤微生物生态学。E-mail: 1044112375@qq.com
