Vorolanib,an oral VEGFR/PDGFR dual tyrosine kinase inhibitor for treatment of patients with advanced solid tumors:An open-label,phase I dose escalation and dose expansion trial
2021-03-13YanSongJinwanWangXiubaoRenJieJinLiMaoChrisLiangLiemingDingLinYang
Yan Song,Jinwan Wang,Xiubao Ren,Jie Jin,Li Mao,Chris Liang,Lieming Ding,Lin Yang
1Department of Medical Oncology,National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100021,China;2 Department of Biotherapy,Tianjin Medical University Cancer Institute and Hospital,Tianjin 300060,China;3 Department of Urology,Peking University First Hospital,Beijing 100034,China;4Betta Pharmaceuticals Co.,Ltd.,Hangzhou 311100,China
Abstract Objective:This study evaluated the safety and preliminary efficacy of vorolanib,a novel tyrosine kinase inhibitor,for treatment of patients with advanced solid tumors.Methods:During dose escalation,patients received increasing doses of oral vorolanib (50-250 mg once daily) in cycles of four weeks for up to one year.During dose expansion,patients received recommended doses (100 and 200 mg) in 4-week cycles.The primary endpoint was to determine the safety and maximum tolerated dose and/or the recommended phase II dose (RP2D).The severity and type of adverse drug reactions (ADRs) were assessed using the Common Terminology Criteria for Adverse Events version 4.0.The second endpoint was preliminary efficacy in terms of objective response and progression-free survival (PFS).Results:No dose-limiting toxicity occurred during dose escalation (50-250 mg).Five (26.3%) patients in the escalation cohort (n=19) and 12 (48.0%) in the expansion cohort (n=25) experienced grade 3 ADRs.The most common ADRs were hair color changes,fatigue,portal hypertension,hypertriglyceridemia,and proteinuria.During dose expansion,the patients treated with 200 mg and 100 mg (once daily) showed an objective response rate of 22.2% and 5.9%,respectively;the disease control rate was 88.9% and 73.3%,respectively;the median PFS was 9.9 [95% confidence interval (95% CI):7.4-not reached]months and 3.8 (95% CI:1.9-not reached) months,respectively.Conclusions:Oral vorolanib at a dose of 200 mg (once daily) exhibited an acceptable safety profile and favorable clinical benefit for patients with advanced solid tumors.The RP2D for vorolanib was determined to be 200 mg as a daily regimen.
Keywords:Vorolanib;TKI;VEGFR;PDGFR;advanced solid tumor
Introduction
Small-molecule receptor tyrosine kinase inhibitors (TKIs)can be used to selectively interfere with receptor tyrosine kinase (RTK) activity,thereby blocking activation of downstream signaling.This protocol represents an effective molecular-targeted therapy for a broad spectrum of advanced cancers (1). Most TKIs are designed to simultaneously target RTKs in multiple signaling pathways.The rationale behind this multi-targeted therapeutic design rests on accumulating evidence that indicates the involvement of multiple signaling pathways in tumorigenesis and the presence of compensatory signaling mechanisms by which resistance emerge (2,3).Among the most targeted RTKs are the vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) families,both of which are major mediators of tumor angiogenesis and provide compensatory signaling for the control of angiogenesis.In addition to its pro-angiogenic effects,the VEGF/VEGFR axis exerts diverse functions as an autocrine regulator of tumor cell function (survival,migration,invasion) and immune suppressor (4).Biallelic inactivation of the von Hippel-Lindau (VHL) gene is a mutational signature in renal carcinogenesis,and occurs in 40%-80% of sporadic renal clear cell carcinoma (RCC) (5).VHLinactivation leads to the overexpression of downstream VEGF and PDGF,thus driving the proliferation of tumor cells and the promotion of tumor angiogenesis by binding to their receptors (6).
The recognition of VEGF and PDGF as key regulators of tumor angiogenesis has led to the development of VEGF-and PDGF-targeted agents that include blocking antibodies to the receptors,DNA or RNA aptamers for ligands,and TKIs.The combined effect on VEGFR and PDGFR,with similar potencies,is critical for the therapeutic efficacy of sunitinib against advanced RCC (7).Sunitinib has also been approved for the treatment of advanced gastrointestinal stromal tumors (GIST) after imatinib progression and for advanced pancreatic neuroendocrine tumors (8,9).This broad spectrum of therapeutic action is thought to be attributable to the angiogenetic targeting of VEGFR and PDGFR and the tumorigenic targeting of c-KIT and FMS-like tyrosine kinase 3 (FLT-3) in sunitinib-based therapies.Another TKI approved as a first-line therapy option for advanced RCC is pazopanib,a second-generation TKI that targets VEGFR,PDGFR,and c-KIT (10).Pazopanib and sunitinib offer similar benefits in efficacy with regards to progression-free survival (PFS),although the safety data appear to favor pazopanib over sunitinib (11).
Vorolanib (also known as CM082) is a novel oral multitargeted TKI that is in the same class as pazopanib and sunitinib and is expected to have similar anti-angiogenic properties based on its specificity against all isoforms of VEGFR and PDGFR.The concept behind the design of vorolanib was based on preclinical pharmacokinetic (PK)and pharmacodynamic (PD) findings that demonstrated that the constant inhibition of VEGFR and PDGFR phosphorylation is not necessary for the anti-tumor efficacy of sunitinib (12).However,sunitinib is eliminated slowly in humans at a plasma half-life of >40 h after a relatively high efficacious dose (13,14).This practice results in constant inhibition when administered using a daily dosing regimen;the actions of this drug are thought to be related to its clinical toxicity.Slow elimination has also been observed for pazopanib,with a terminal plasma half-life of approximately 31 h in humans (15).Therefore,vorolanib was designed to meet the needs of intermittent inhibition to minimize potential toxicity while maintaining a therapeutic efficacy that is similar to that of sunitinib.Preclinical PK data have revealed that vorolanib is associated with good oral bioavailability,high plasma protein binding and more importantly,fast clearance and low organ accumulationin vivo(unpublished data).Clinical PK data have also indicated that vorolanib has a relatively short plasma half-life of 4-8 h without any obvious accumulation in humans (16).Preclinical toxicity data have further indicated that vorolanib is well tolerated when administered in a single-dose administration although dose dependency and reversible toxicity may occur with multiple dosing (unpublished data).Based on these preclinical and clinical data,we conducted a phase I clinical study to assess the safety and preliminary efficacy of vorolanib in patients with advanced solid tumors.
Materials and methods
Study design and patients
This single-arm phase I study consisted of a dose-escalation and dose-expansion phase.The primary objective was to evaluate the safety and tolerability of vorolanib in patients with documented advanced solid tumors.The doseescalation phase was conducted at the Cancer Hospital of Chinese Academy of Medical Sciences.The dose-expansion phase was conducted at three sites (Cancer Hospital of Chinese Academy of Medical Sciences,Peking University First Hospital,and Tianjin Medical University Cancer Institute and Hospital). The study protocol and amendments were reviewed and approved by each local Ethics Committee (Supplementary Table S1). Studies involving human participants were reviewed and approved by the Ethics Committee of Cancer Hospital,Chinese Academy of Medical Sciences.All patients provided written informed consent prior to commencing any protocolrelated procedures.The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice.Eligible patients were 18-65 years old;and had histologically or cytologically confirmed RCCs,GISTs,or other advanced solid malignancies (aged 18 years or older and diagnosed with histologically or cytologically confirmed RCCs in the dose-expansion phase).Other eligibility criteria for both phases included a body mass index (BMI) of 18-28 kg/m2;a measurable disease according to the Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1);a disease that was not responsive to standard therapies or for which there was no effective therapy;patient refusal for other recommended therapies; an Eastern Cooperative Group (ECOG)performance status of 0-1;a life expectancy ≥12 weeks;adequate bone marrow function (an absolute neutrophil count ≥1.5×109/L,a platelet count ≥100×109/L,and a hemoglobin concentration ≥100 g/L);and adequate major organ function [total bilirubin ≤1.5× the upper limit of normal (ULN),alanine aminotransferase and aspartate aminotransferase ≤1.5× ULN if no liver involvement or ≤2.5× ULN with liver involvement,and a serum creatinine ≤1.5× ULN].Patients were excluded from the trial if they had received anti-tumor therapies (i.e.,chemotherapy,radiotherapy,immunotherapy,or hormonal therapy)within 4 weeks or major surgery within 12 weeks of enrollment;if they were experiencing residual toxicity from previous therapies or had not fully recovered from prior surgery;if they had been administered with strong cytochrome P450 3A4 inducers or inhibitors within the previous 2 weeks;if they had concomitant medications that could prolong the corrected QTc interval and/or torsades de pointes;or if they had a history of hypersensitivity to VEGFR TKIs (e.g.,sunitinib,sorafenib,pazopanib).Patients were also excluded if they were pregnant or breastfeeding;if they were unwilling to use contraception;if they were experiencing symptomatic brain metastases or active gastrointestinal diseases that precluded the absorption,distribution,metabolism,or excretion of medication;if they had hepatitis B virus infection;if they were immunodeficient;if there was any history of substance abuse,or if there were any psychiatric or medical conditions that may affect compliance to the study.This trial was registered at ClinicalTrials.gov (No.NCT01863485).
Procedure
The treatment regimens are shown inSupplementary Figure S1A.In the dose-escalation phase,a single dose of vorolanib(CM082 tablet,Challenge Meditech,Shanghai,China) was administered orally.If no dose-limiting toxicity (DLT)occurred,the patient then received oral vorolanib at this dose level on a daily basis for 4 consecutive weeks (cycle 1).If no DLT occurred during cycle 1,then treatment was continued (on a 4-week cycle) until disease progression,unacceptable toxicity,non-compliance,or the patient withdrew consent.The starting dose was 50 mg and was administered orally once daily;this dose was escalated to 100,150,200,and 250 mg in sequential cohorts of 3-6 patients using a standard dose escalation design.The starting dose and dose range were determined by preclinical toxicological studies performed in rats and dogs(the lowest observable adverse effect concentration was 9,318.8 ng·h/mL in male rats,104,474.5 ng·h/mL in female rats,4,102.7 ng·h/mL in male dogs,and 4,834.6 ng·h/mL in female dogs).We also considered the safety/tolerability data provided by a US-based phase I study,in which vorolanib was well tolerated.Only grade 1 toxicity was encountered in patients receiving oral vorolanib at a daily dose range of 50-150 mg when fasting or after breakfast.DLT was defined as occurring between the first dose and the end of the first cycle in any of the following circumstances:grade 4 hematologic toxicity or febrile neutropenia,≥grade 3 hepatotoxicity,≥grade 3 nonhematologic toxicity,and ≥grade 2 renal toxicity.Dose escalation proceeded when a minimum of three patients completed the first cycle with no DLT at a given dose level.If one patient experienced DLT,three additional patients were treated at that dose level;dose escalation was only applied if there were no additional DLTs.If more than two patients experienced DLTs,then dose escalation was terminated and the dose level immediately below this level was determined to be the maximum tolerated dose(MTD).Intra-patient dose escalation was not permitted.However,dose interruption and reduction were allowed according to the severity of hematological and nonhematological toxicities.Dose expansion proceeded with two dose levels of 100 and 200 mg once daily.During dose expansion,a single dose of oral vorolanib was administered.This was then followed by one day off treatment and 4-week cycles of once-daily vorolanib until disease progression or unacceptable levels of toxicity.
Safety evaluations [vital signs,physical examination,ECOG performance status,hematology,blood chemistry,urinalysis,and 12-lead electrocardiograms (ECGs)]were performed for all patients at baseline and then at regular intervals thereafter.Disease evaluation was conducted at baseline and every eight weeks.Tumor response was categorized according to RECIST 1.1,using contrastenhanced computed tomography (CT) or other imaging modalities.
Outcomes
The primary endpoint of this study was to assess the safety and determine the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) for vorolanib.Safety assessment was based on the incidence,severity,and types of adverse events (AEs).The severity of AEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE)version 4.0.An AE related to the protocol treatment was defined as an adverse drug reaction (ADR).The second endpoint was the assessment of treatment efficacy.This included the best percentage change from baseline in target lesion measurements,objective response rate [ORR,defined as the proportion of patients who achieved complete response (CR) and partial response (PR),in accordance to RECIST 1.1],disease control rate (DCR,defined as the proportion of patients who achieved CR,PR,and SD),time to progression (TTP,defined as the time from the first dose to tumor progression),and PFS (defined as the time from the first dose to disease progression or death from any cause).Objective responses (CR or PR)were confirmed by one sequential tumor assessment at least four weeks later.
Statistical analysis
Descriptive statistics were used to calculate the mean,standard deviation (SD),median,interquartile range,and frequency;these parameters allowed us to summarize baseline characteristics and safety data.Safety was analyzed based on the safety set (SS),which included all patients who received at least one dose of vorolanib.The primary efficacy analysis was based on the full analysis set (FAS),which included all patients in the FAS except for those who received only the first dose or had no valid measurements for efficacy outcomes.Additionally,efficacy was analyzed in the per-protocol set (PPS);this included all patients in the FAS without major deviations.TTP and PFS were analyzed using the Kaplan-Meier method,including medians and corresponding 95% confidence intervals (95%CIs). Statistical analyses were performed using SAS software (Version 9.1.3;SAS Institute Inc.,Cary,NC,USA).
Results
Patient disposition and baseline characteristics
Between May 2013 and August 2014,19 out of 26 screened patients with advanced solid tumors were enrolled and treated in the dose-escalation phase;the last patient completed the phase on 3rd March,2015.Between June 2014 and May 2015,25 of the 27 screened patients with advanced RCC were enrolled in the dose-expansion phase;the last patient completed this phase on 25th May,2016.Supplementary Figure S1Bshows a flow chart depicting patient allocation from the initial screening to the final analysis.All patients in both phases received at least one dose of vorolanib and were included in the SS.The FAS consisted of 17 patients in the dose-escalation phase and 24 patients in the dose-expansion phase after excluding patients who withdrew from the study before completing cycle 1.The PPS consisted of 15 and 20 patients in the escalation and expansion cohorts,respectively.
Baseline patient characteristics are summarized inTable 1.The median age at enrollment was 50 (range,22-63) years and 60 (36-76) years in the escalation and expansion cohorts,respectively.The majority of patients in both phases was male and had an ECOG performance status score of 1.The majority (63.2%) of the escalation cohort,and all of the expansion cohort,had a primary diagnosis of RCC.All patients had stage IV disease.Surgery was performed for the majority of patients prior to this trial commencing.Of note,11 (57.9%) and 14 (56.0%)patients in the escalation cohort and expansion cohort,respectively,had previously been treated with anti-VEGF agents (e.g.,sorafenib,pazopanib,erlotinib,bevacizumab,and recombinant human endostatin).
Safety and tolerability
In the dose-escalation phase,19 patients were available for safety analysis and had a median exposure duration of 120(range,1-373) d.No DLT was seen across any of the dose cohorts;consequently,MTD was not reached.The number of AEs and ADRs increased with dose across the range of 50-150 mg.Patients treated at the maximum-administered dose (MAD) experienced more grade 3 AEs and ADRs compared with the other dose groups.Three (60.0%) of five patients treated at MAD had grade 3 AEs,compared to 50.0% (2/4 patients) in the 100 mg group and 33.3% (1/3 patients) in the 150 and 200 mg groups.None of the patients in the 50 mg group had grade 3 AEs.Similarly,grade 3 ADRs in 50-250 mg groups were encountered by 0,0,33.3%,33.3% and 60.0% of patients in the 50-250 mg groups,respectively.However,patients treated at MAD experienced less AEs [1/5 (20.0%) patients]and ADRs [1/5(20.0%) patients] that needed medical interventions,compared with those in 50 mg group [1/4 (25.0%) patients,and 1/4 (25.0%) patients],100 mg group [3/4 (75.0%)patients,and 1/4 (25.0%) patients],150 mg group [2/3(66.7%) patients,and 2/3 (66.7%) patients]and 200 mg group [2/3 (66.7%) patients,and 2/3 (66.7%) patients].All patients experienced at least one AE or ADR (Table 2).A total of 14 grade 3 AEs occurred in 7 (36.8%) patients.Of these,7 events in 5 (26.3%) patients were considered to be ADRs;no grade 4 AE occurred.Two patients discontinued treatment due to drug-related AEs (one grade 2 ischemic stroke in the 50 mg cohort,and one grade 3 urinary retention in the 100 mg cohort).Dose interruption due to AEs occurred in 3 (15.8%) patients;two of these patients were subsequently administered a reduced dose.Common ADRs (with an incidence ≥20%) are listed inTable 2.The most common ADRs (for any grade) were hair color changes,hypertriglyceridemia and proteinuria;these ADRs were recorded in 15 (78.9%),12 (63.2%),and 12 (63.2%)patients,respectively.Based on safety data derived from the escalation cohort,we selected 100 and 200 mg (once daily)as the doses to be used in the expansion phase.Twenty-fivepatients were available for safety assessment with a median exposure duration of 196 (range,39—367) d (Supplementary Figure S1B).All patients experienced at least one AE/ADR(Table 2).The most common ADRs were hair color changes (17,68.0%),fatigue (16,64.0%) and portal hypertension (13,52.0%).Fourteen (56.0%) patients experienced 24 grade 3 or higher AEs,17 of which were ADRs;these occurred in 12 (48.0%) patients.None of these ADRs were grade 4.Eight serious AEs occurred in 4(16.0%) patients.Only one (grade 3 neutropenia with fever) was considered to be related to the treatment,and was reported in a patient treated once daily at 100 mg.One(4.0%) patient discontinued treatment due to an ADR,which was a grade 2 palpitation.Treatment interruption due to AEs occurred in 4 (16.0%) patients;two of these patients required a dose reduction.In terms of group differences,the incidences of grade 3 ADRs were comparable between the 100 and 200 mg (once daily)groups (46.7%vs.50.0%).AEs requiring treatment occurred in 80% of patients treated with once-daily 200 mg vorolanib,which was higher than the proportion (66.7%)in the group receiving 100 mg.However,AEs leading to dose interruption or reduction [1/10 (10.0%)vs.3/15(20.0%)]or discontinuation [0 (0%)vs.1/15 (6.7%)]were less frequent in the 200 mg group than in the 100 mg group.
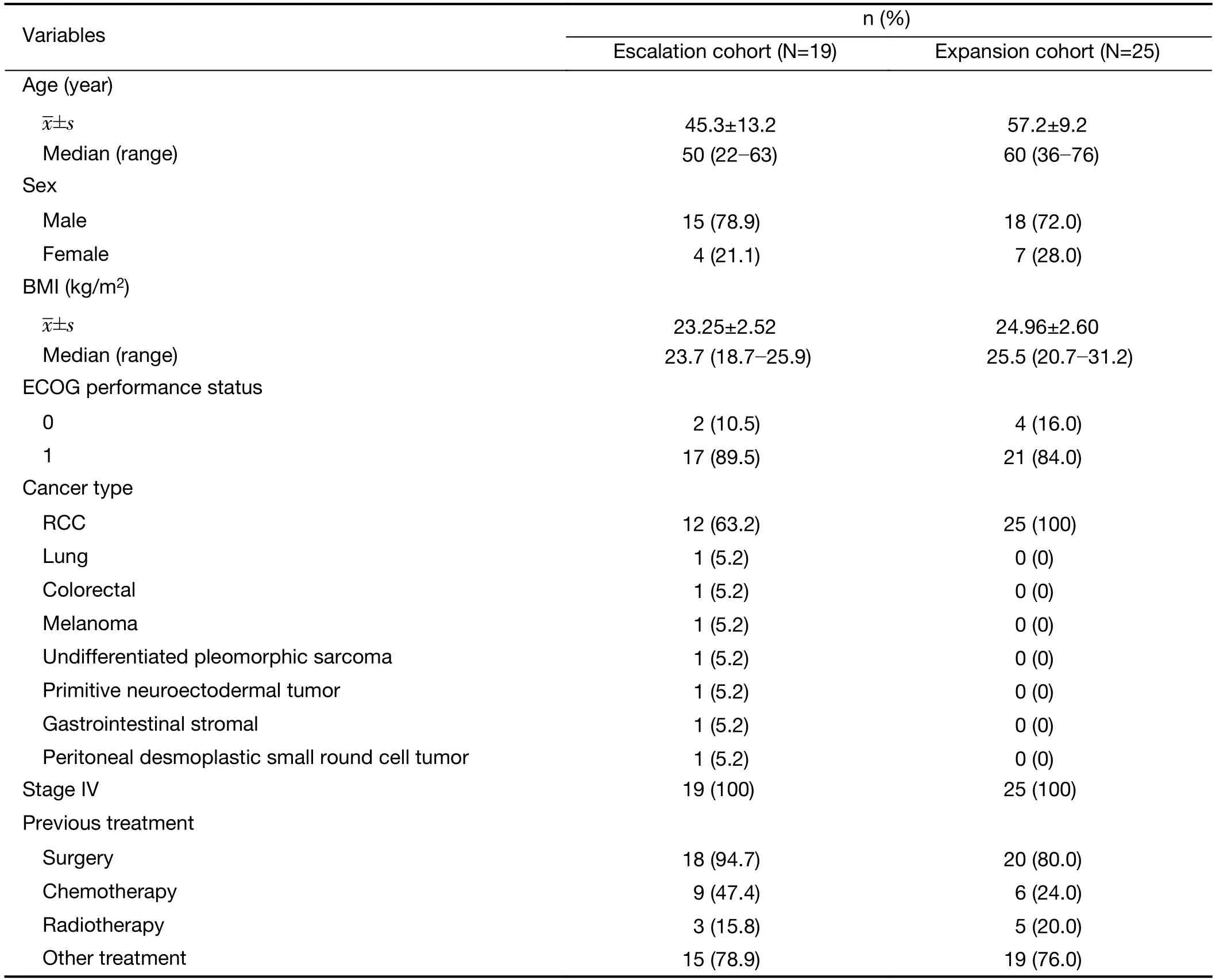
Table 1 Baseline patient characteristics
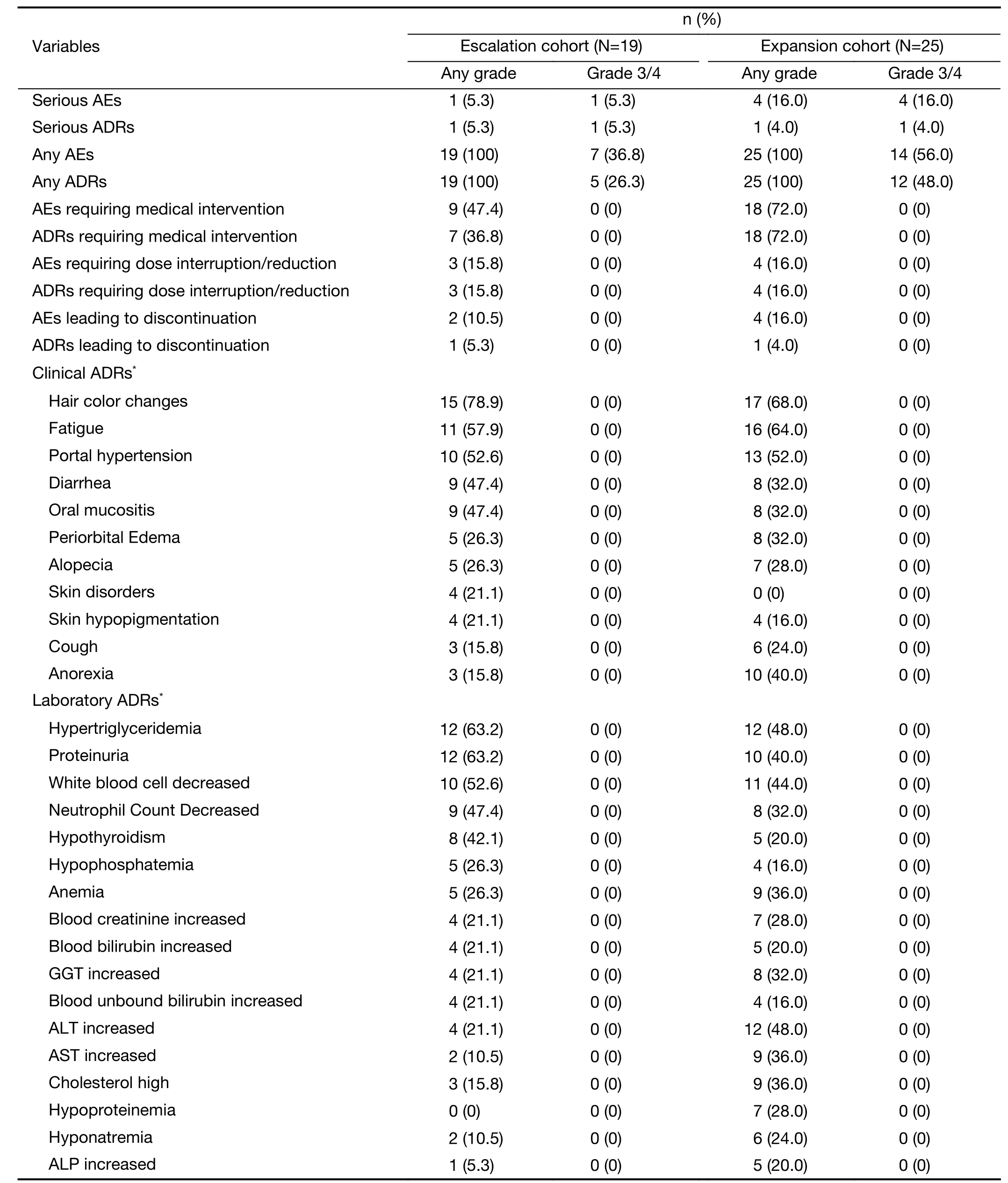
Table 2 Safety evaluation
Clinically relevant differences from baseline or consistent trends were observed for several other laboratory parameters (blood chemistry,hematology and urinalysis),vital signs and ECG.In general,the most frequently reported hematological and blood chemical abnormalities in both phases were reductions in leukocytes,neutrophils,hemoglobin and platelets;and elevations of bilirubin (total,bound,and unbound),alanine aminotransferase (ALT),aspartate aminotransferase (AST),creatinine,triglyceride and cholesterol from baseline.The most common abnormalities emerging from urinalysis after baseline included positive urinary protein,erythrocytes and glucose.During dose escalation,sinus rhythm and premature ventricular contraction in ECG signals were reported as a grade 1 AE that could have been related to treatment in one (5.3%) patient.During dose expansion,abnormal ECG measurements were reported in 7 (28.0%) patients who had normal ECGs at baseline.Of these,one patient in the group receiving 100 mg (once daily) experienced grade 1 QTc interval prolongation.
Efficacy
During the dose-escalation phase,17 patients in the FAS available to investigate specific responses and the objective responses are summarized inTable 3.In the escalation cohort,the ORR was 5.9% (1/17) and DCR was 64.7%(11/17).Among 17 patients,12 RCC patients (PR:1;SD:9;PD:2) in the escalation cohort exhibited an ORR and DCR of 8.3% and 83.3%,respectively.During dose expansion,24 patients in the FAS were available for efficacy analysis.Tumor shrinkage was observed in 14 patients (100 mg:n=7;200 mg:n=7;Figure 1).A higher ORR was observed in the group receiving the 200 mg dose (22.2%)when compared with that in the group receiving the 100 mg dose (6.7%) (Table 3);DCR was similar whencompared across the two dose cohorts (100 mg:73.3%;200 mg:88.9%).Figure 1displays the best percentage change in tumor size;one patient was excluded fromFigure 1because there was no measurable target lesion at baseline.Figure 2shows representative CT images for a patient achieving PR in the dose-expansion cohort.
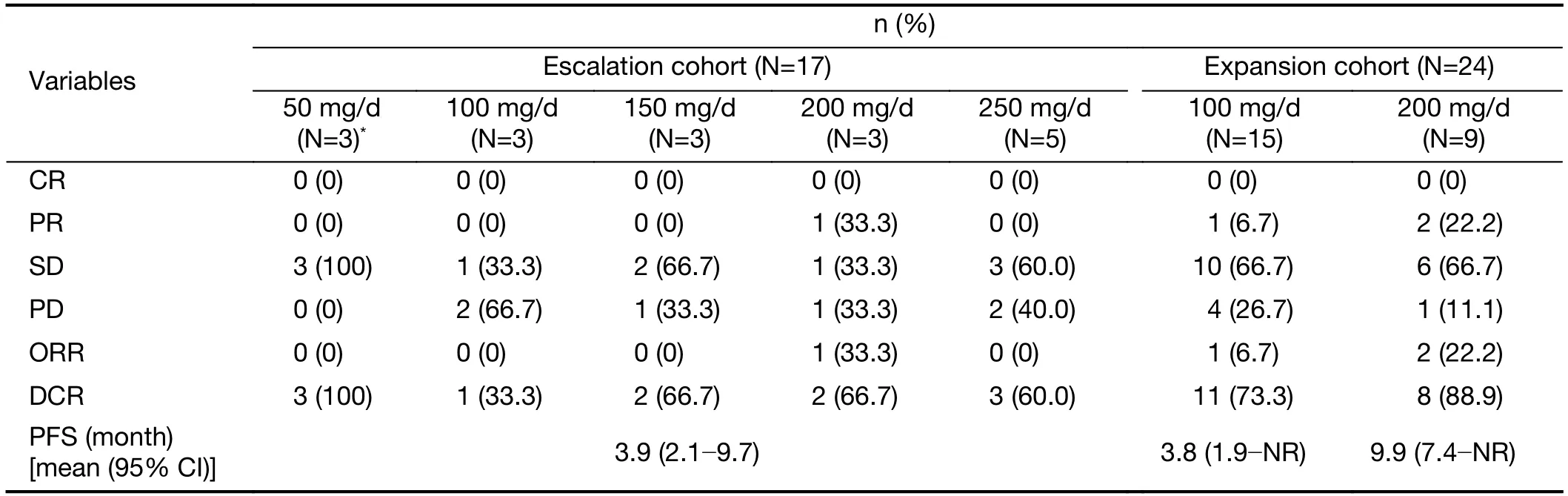
Table 3 Best tumor response and PFS

Figure 1 Waterfall plots for the best percentage change from baseline with regards to measurement of target lesion.Each bar represents an individual patient.
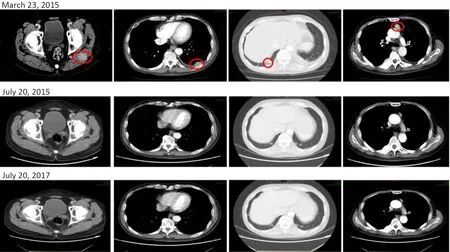
Figure 2 CT scans for one patient achieving PR in dose-expansion cohort before and after vorolanib treatment.This patient was diagnosed with RCC in 2010 and had been treated with axitinib between January 2011 and September 2015 until progression.Baseline imaging (23rd March,2015) indicated metastasis to soft tissue in the left hip and left chest,right lower lobe,and mediastinal lymph nodes.The patient was enrolled and treated with vorolanib on the 31st March,2015.Tumor shrinkage was observed on the July 20th,2015 and July 20th,2017.PR was achieved at the first follow-up visit (25th May,2015).PR had been sustained up to the most recent follow-up visit (20th July,2019).CT,computed tomography;PR,partial response;RCC,renal clear cell carcinoma.
In PPS,15 patients in the FAS were available to evaluate response.In the escalation cohort,the ORR was 6.7%(1/15) and the DCR was 60.0% (9/15).During dose expansion,20 patients in the PPS were available for efficacy analysis.The ORR and DCR were higher in the 200 mg group (ORR:22.2%;DCR:88.9%) than in the 100 mg group (ORR:9.1%;DCR:63.6%) (Supplementary Table S2).
At the time of the data cut-off (25th May,2016),PFS for the escalation cohort was 3.9 (95% CI:2.1-9.7) months.The median PFS for the expansion cohort were 7.4 (95%CI:3.7-not reached) months.The median PFS for the 100 and 200 mg groups was 3.8 (95% CI:1.9-not reached)months and 9.9 (95% CI:7.4-not reached) months,respectively (Figure 3).
Discussion
Preclinical studies have indicated that vorolanib has a shorter half-life than approved TKIs for advanced RCC sunitinib and pazopanib.Furthermore,vorolanib is known to exert substantial inhibitory effects on VEGFR,PDGFR,c-KIT,and FLT-3,thus leading to the inhibition of vascular endothelial cell proliferation and lumen formationin vitro(16,17).The inhibition of VEGF signaling has been shown to induce the regression of choroidal neovascularization in a rat model treated with vorolanib(17).A subsequent phase I dose-escalation trial in patients with neovascular age-related macular degeneration showed a favorable toxicity profile when administered with a daily dose of 200 mg of vorolanib in the vast majority of patients who completed 24-week treatment reported maintained or improved visual acuity (18).As an angiogenesis inhibitor,vorolanib demonstrated broad-spectrum anti-tumor activity in preclinical studies that incorporated murine xenograft models.These positive preclinical findings led to the clinical evaluation of vorolanib in patients suffering from advanced solid tumors.In this phase I clinical study,vorolanib demonstrated a tolerable safety profile with few grade 3/4 AEs in patients with advanced solid tumors.Our study cohorts were predominantly advanced RCC patients who had responded poorly to standard therapies or for whom no effective therapy existed.The RP2D was determined as 200 mg (once daily) with continuous administration.Tumor responses were only observed in patients with advanced RCC,partly due to the fact that we had inadequate numbers of patients with other types of solid tumors.Our results are consistent with results arising from a previous phase I study that reported the excellent tolerability of vorolanib when combined with everolimus in patients with advanced RCC;this study recommended a regimen consisting of 200 mg vorolanib and 5 mg of everolimus as the RP2D (19).

Figure 3 Progression-free survival in (A) Dose-escalation cohort and (B) Dose-expansion cohort.
We found that vorolanib was generally well tolerated in patients with advanced solid tumors when administered once daily at a dose of 50-250 mg.Most AEs were mild or moderate in severity and did not lead to discontinuation of treatment.The most commonly reported ADRs were hair color changes,fatigue,hypertension,hypertriglyceridemia and proteinuria.These AEs appear to be a generalized effect associated with angiogenesis inhibitors targeting the VEGFR family (20,21).This safety profile was consistent with that observed in a previous phase I trial of vorolanib for solid tumors,in which the vast majority of patients were Caucasians and the daily doses tested ranged from 50 to 800 mg (16).In this study,grade 3 ADRs were recorded in 38.6% of patients and no grade 4 ADRs were observed.ADRs that caused dose interruption/reduction occurred in 17.1% of patients while those that caused discontinuation were reported in 4.5% of patients.The frequencies of ADRs appeared to be lower than those reported in sunitinib-and pazopanib-treated patients.In a phase III trial of sunitinib in patients with metastatic RCC,grade 3/4 ADRs were recorded in 77% patients;52% patients experienced dose reduction related to ADRs and 20%patients were discontinued due to ADRs (22).Similar frequencies were observed for patients treated with pazopanib;dose reduction and discontinuation due to ADRs were reported in 44% and 20% of patients,respectively,in a phase III trial for metastatic RCC (11).In a previous phase III trial that compared axitinib and sorafenib as second-line TKIs for metastatic RCC,55%and 62% of patients in the axitinib and sorafenib groups received dose interruption or reduction,respectively.The proportions of patients who were discontinued due to ADRs arising from axitinib and sorafenib treatment were 9% and 13%,respectively (23).Given the relatively small number and heterogenous nature of the patients enrolled in the present study,it is too early to ascertain whether vorolanib is superior to other VEGFR/PDGFR dual TKIs in terms of safety.In order to reach a more definitive conclusion,it will be necessary to carry out large phase II trials and head-to-head trials that compare safety profiles between vorolanib and other approved TKIs.For now,it is reasonable to argue that the safety profile of vorolanib appears favorable if not superior to that of other TKIs when used to treat advanced RCC.
One primary objective of this clinical study was to determine the MTD and/or RP2D for vorolanib.Because no DLTs were observed within the first cycle of the doseescalation phase,no MTD was established and the MAD was defined as 250 mg.This is in agreement with a previous study of vorolanib that reported no DLTs in patients with advanced solid tumors tum up to a dose of 800 mg.Further escalation was not attempted because apparent saturation of absorption was observed at doses of 400-800 mg (16).In the present study,the frequencies of AEs/ADRs increased with increasing dose within a range of 50-150 mg,and reached a plateau at 150 mg and greater.Therefore,following the escalation phase,we selected 100 and 200 mg as the doses of vorolanib to use during the expansion phase.There was no difference in the overall safety profile when compared between these two doses.AEs leading to dose interruption,dose reduction,and discontinuation were less frequent in the 200 mg group than in the 100 mg group.One possible reason for this was the small sample size.
In the present clinical trial,we observed tumor responses to vorolanib in patients with advanced solid tumors.Four(9.8%) of the patients that were available for analysis achieved PR;all four patients diagnosed with stage IV RCC and three of these PRs occurred in patients treated with a dose of 200 mg (once daily).SD was the best response in patients with non-RCC tumors,indicating limited antitumor activity,although the number of patients was too small for us to arrive at any meaningful conclusion regarding these tumor types.Of the 25 patients with RCC in the dose-expansion phases,the ORR was 5.9% in the 100 mg group and 22.2% in the 200 mg group.One third of patients in the 100 and 200 mg groups achieved SD within one year of treatment;the DCR was 73.3% and 88.9%,respectively.Importantly,disease control was prolonged in patients when treated daily with 200 mg of vorolanib;the median PFS of 9.9 months in the 200 mg group was considerably longer than the median TTP of 3.8 months in the 100 mg group.The duration of exposure in 200 mg group and 100 mg group was 265 (range,39-367)d and 114 (range,56-367) d,respectively.The ORR of 22.2% and the median PFS of 9.9 months in patients treated with 200 mg of vorolanib were comparable with the results of a recent meta-analysis that reported a pooled ORR of 27.9% (95% CI:24.2-32.0) and median PFS of 9.3(8.6-10.2) months for sunitinib in patients with metastatic RCC (24).Notably,this study enrolled patients with advanced solid tumors who had not responded well to standard therapy;91% of these patients had been previously treated with systemic therapy and 59% were previously treated with anti-angiogenic therapy.Therefore,it is possible that the efficacy of vorolanib had been underestimated and further investigation of vorolanib in untreated patients in order to justify the potential of vorolanib as a first-line therapy was warranted.
This study had important strengths and certain limitations that need to be considered.Despite the small sample size,our results are consistent with the preclinical hypothesis and expectation that vorolanib is well tolerated and associated with lower toxicity than other TKIs while maintaining similar levels of anti-tumor activity and a long plasma half-life.Patients were offered vorolanib therapy that continued for a maximum of 13 cycles within a 1-year period unless disease progression or intolerable toxicity occurred.This was longer than the median overall survival of 10 months in patients with advanced renal cancer (25).Because this was a relatively long trial period,there was a reduced possibility of missing information relating to the long-term AEs and efficacy of vorolanib therapy.The main limitations of the present study were the small sample sizes available for each phase and the lack of non-RCC tumor types;these factors limited the strengths and generalizability of our conclusions.
Conclusions
A 200 mg once-daily dose of vorolanib was associated with an acceptable safety profile and achieved favorable clinical benefits that were consistent with what would be expected for a VEGFR/PDGFR dual TKI for advanced solid tumors,especially for patients with RCC.A 200 mg dose(once daily) was determined as the RP2D for further assessment in phase II studies.Additional studies are now in progress to further explore the clinical benefits of vorolanib as a single agent and in combination with other agents of targeted therapy.These studies aim to validate the findings from our phase I trial.
Acknowledgements
None.
Footnote
Conflicts of Interest:Li Mao,Chris Liang,and Lieming Ding were employed by Betta Pharmaceuticals Co.,Ltd.No potential conflicts of interest were disclosed by the other authors.
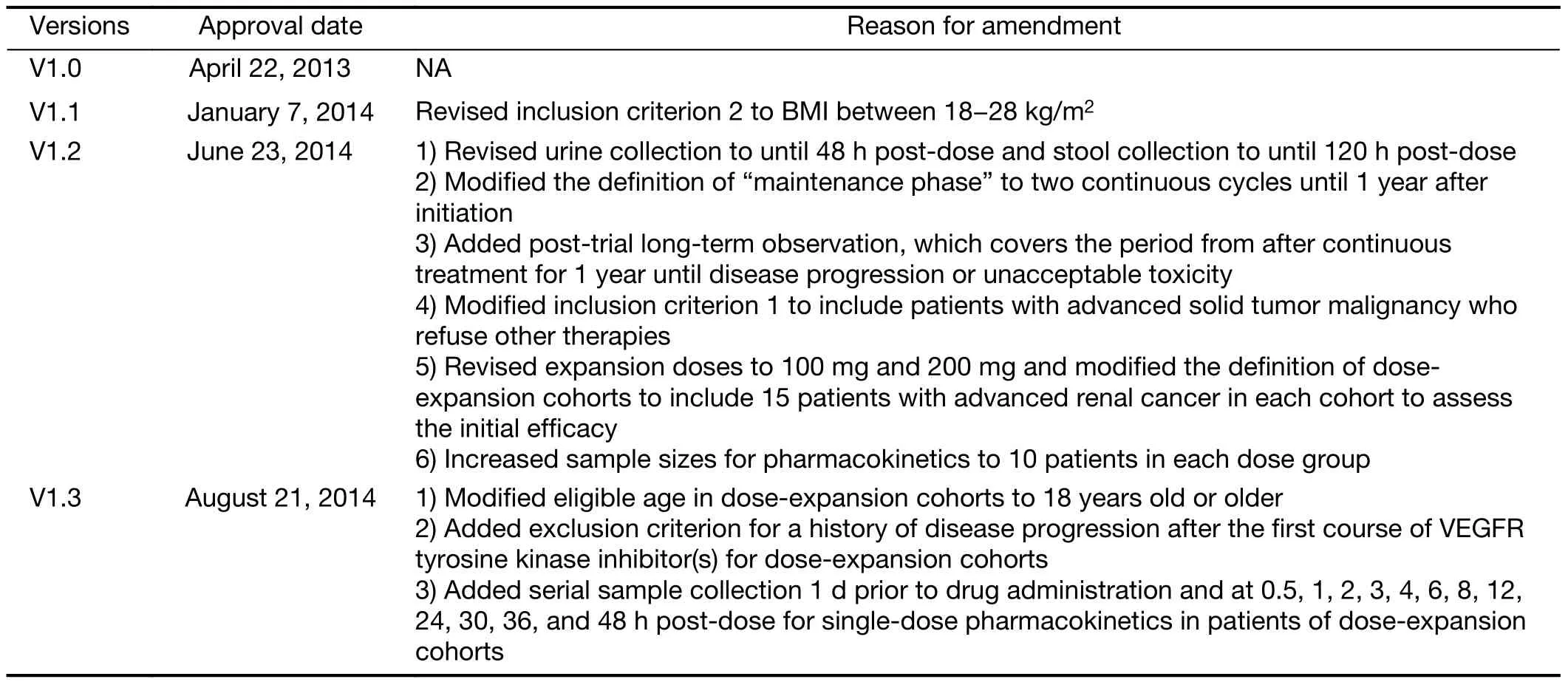
Table S1 Protocol amendments

Table S2 Best tumor response

Figure S1 Trial profile.(A) Treatment regimens;(B) Diagram depicting patient allocation.AE,adverse effect;SS,safety set;FAS,full analysis set;PPS,per-protocol set.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Colorectal cancer burden and trends:Comparison between China and major burden countries in the world
- Changing trends of disease burden of gastric cancer in China from 1990 to 2019 and its predictions:Findings from Global Burden of Disease Study
- Clinical characteristics and clinicopathological correlations of bilateral breast cancer in China:A multicenter study from Chinese Society of Breast Surgery (CSBrS-006)
- Multi-center investigation of breast reconstruction after mastectomy from Chinese Society of Breast Surgery:A survey based on 31 tertiary hospitals (CSBrS-004)
- Laparoscopic vs. open surgery for gastrointestinal stromal tumors of esophagogastric junction:A multicenter,retrospective cohort analysis with propensity score weighting
- Radiotherapy combined with nimotuzumab for elderly esophageal cancer patients:A phase II clinical trial