非金属元素N-S共掺杂SnO2电性能第一性原理研究*
2021-03-08李昊天王景芹孙绍琦梁雨婷朱艳彩
李昊天,王景芹,孙绍琦,张 哲,梁雨婷,朱艳彩
(1.河北工业大学省部共建电工装备可靠性与智能化国家重点实验室,天津 300130;2.河北工业大学河北省电磁场与电器可靠性重点实验室,天津 300130)
0 引 言
AgCdO长久以来被广泛被称作为“万能”的电接触材料。然而,AgCdO在工作中会产生的Cd蒸汽不仅仅对人有毒性,也对环境造成很大的污染,这种“万能”的触头材料逐渐被淘汰,人们转而去寻找替代品[1]。
随着研究的发现,AgSnO2被发现了具有良好的耐磨性、耐腐蚀以及使用寿命很长,有望成为替代AgCdO的材料。但是,AgSnO2实际上使用还存在着许多问题。由于工作环境温度高,触头表面就会渗出SnO2。SnO2作为宽禁带半导体材料,导电性能并不理想,这也就导致了AgSnO2使用时的电性能较差[2-3]。所以就有必要对SnO2进行改性。目前已知的对SnO2改性的方法主要包括对其进行掺杂不同的元素,来提升SnO2的物理性质。截止至现在,已经有非常多的学者在利用不同种的元素对AgSnO2进行掺杂并取得了不小的成果。
非金属元素(C、N、S、F等)在自然界中贮存量非常的大,价格便宜,方便易得。关于掺杂不同元素的研究已经取得不少成果,徐剑等确定了F掺杂对O的替换位置,掺杂后的体系态密度存在向低能方向的移动,费米能级进入导带,能级发生简并,载流子浓度增加,价带宽度缩小,而导带展宽,禁带宽度变窄[4];彭彩云等采用第一性原理密度泛函理论框架下的平面波超软赝势方法分别建立在SnO2中单掺杂不同浓度的C元素,发现SnO2的带隙随着掺杂C浓度的增加而减小,在浓度为16.7%时带隙最小[5];刘周等利用La-S共掺杂SnO2,发现共掺杂体系的稳定性随着S原子的浓度增大而下降,共掺杂可以改善SnO2的导电性,并且随着S原子浓度的增大,导电性进一步增强[6];康慧玲等发现SnO2共掺杂Sr、F贡献了部分电子,使共掺杂SnO2产生带负电的晶体缺陷,因此改善了SnO2的导电性能[7]。然而关于非金属共掺大多都是光电性能的研究,对SnO2非金属的掺杂的电性能研究成果鲜有。本文利用N和S两种非金属元素对SnO2进行共掺杂,使用第一性原理计算方法对掺杂非金属后的晶胞进行优化和模拟仿真运算,分别分析优化后共掺杂不同浓度的N和S的SnO2的能带结构,态密度、电荷布局数等电性能,为AgSnO2触头材料研究提供新思路和理论依据。
1 SnO2模型与计算方法
1.1 理论模型
本文以理想的金红石结构SnO2作为研究对象,其空间群为P42/MNM(No.136),属于四方晶系,晶格常数a=b=0.4830 nm,c=0.3236 nm,α=β=γ=90°[8],单个SnO2晶胞当中含有2个Sn原子和4个O原子。本文采用1×2×3超晶胞36个原子,包括12个Sn原子和24个O原子。利用N和S替换O原子,选择掺杂浓度为16.7%,掺杂模型如同1所示:

图1 掺杂结构模型
1.2 计算方法
本文利用基于密度泛函理论的第一性原理方法,用MaterialsStudio软件当中的CASTEP模块对本征SnO2、N-SnO2、S-SnO2、以及N-S-SnO2体系当中的晶体结构和电学性质进行计算研究。计算过程在倒易空间中进行,使用基于密度泛函理论的第一性原理方法,采用广义梯度近似(Generalized Gradient Approximation,GGA)中的PBE计算。平面波截断能量选取为340 eV,布里渊区K网格选为3×2×1,利用BFGS(Broyden Flecher Goldfarb Shanno)算法对各掺杂结构进行几何优化,迭代收敛精度1.0×10-6eV/atom,应力偏差不大于0.05GPa,原子间相互作用力低于0.3eV/nm[9]。在开始计算前,要先对每个晶体体系进行几何优化,得到合理的稳定结构晶体,再对其进行电性能分析。选用价电子组态:Sn(5s25p2),O(2s22p4),N(2s22p3),S(3s23p4)。
2 结果讨论
2.1 不同掺杂系统的稳定性分析
优化后的掺杂系统焓变的大小决定了体系的热力学稳定,其对应得数值的绝对值越大,说明该体系越稳定,计算数值如表1所示。由表中数据不难观察出,掺杂前后体系焓变小于本征SnO2的体系焓变,这说明优化体系均处于热力学稳定状态。而且共掺杂优化体系的焓变绝对值最大,这就意味着热稳定性最好。

表1 各掺杂体系焓变值
2.2 能带结构
本征SnO2以及各个掺杂浓度体系的SnO2的能带结构如图,选择0eV为费米能级。观察图像可以发现掺杂前后的导带底和价带顶在布里渊区的同一位置,说明SnO2掺杂前后优化体系均为直接带隙半导体。特别的,可以从图2(a)中观察到,本征SnO2的带隙计算值为1.36eV,和实验值3.6eV差距比较明显,这主要是在仿真计算过程中,对于由于广义梯度近似法计算具有一定的局限性,低估导带中激发态电子的能量[10],产生一定的误差。不过由于是对比不同掺杂体系的带隙相对大小,所以所产生的误差对于结果影响不大。掺杂后,单掺和共掺与本征结构相比能带曲线均密集变化,导带的部位变窄,向低能方向移动,局域性增强,在导带中的电子有效质量相对增大。
图2(b)为S单掺杂体系的能带结构,相较于本征SnO2,由于单掺S属于p型掺杂, 费米能级没有进入导带, 而是处于价带顶的杂质能级中,也观察到了杂质能级,使带隙值减小到0.52eV[11]。图2(c)为N元素单掺杂体系的能带结构,可以观察到在-12.5eV处有新的杂质能级,杂质能级的引入使得导带底和价带顶均发生变化,N原子2p能级进入带隙,N原子的掺杂也使得与之最近邻的O和Sn的电子结构受到影响,在相同能量范围产生s电子和p电子,N、O、Sn轨道能级重叠,耦合作用增强[12-13],带隙减小到了0.34eV,载流子浓度增加,导电性增强。图2(d)为N-S共掺杂的能态图,带隙进一步减小到为0.26eV,价带顶被S原子的3p轨道和N原子,2p轨道占据,会有更多的空穴载流子,价带顶部向上移动。电子跃迁所需要的能量更加的少了,理论上更加容易导电。
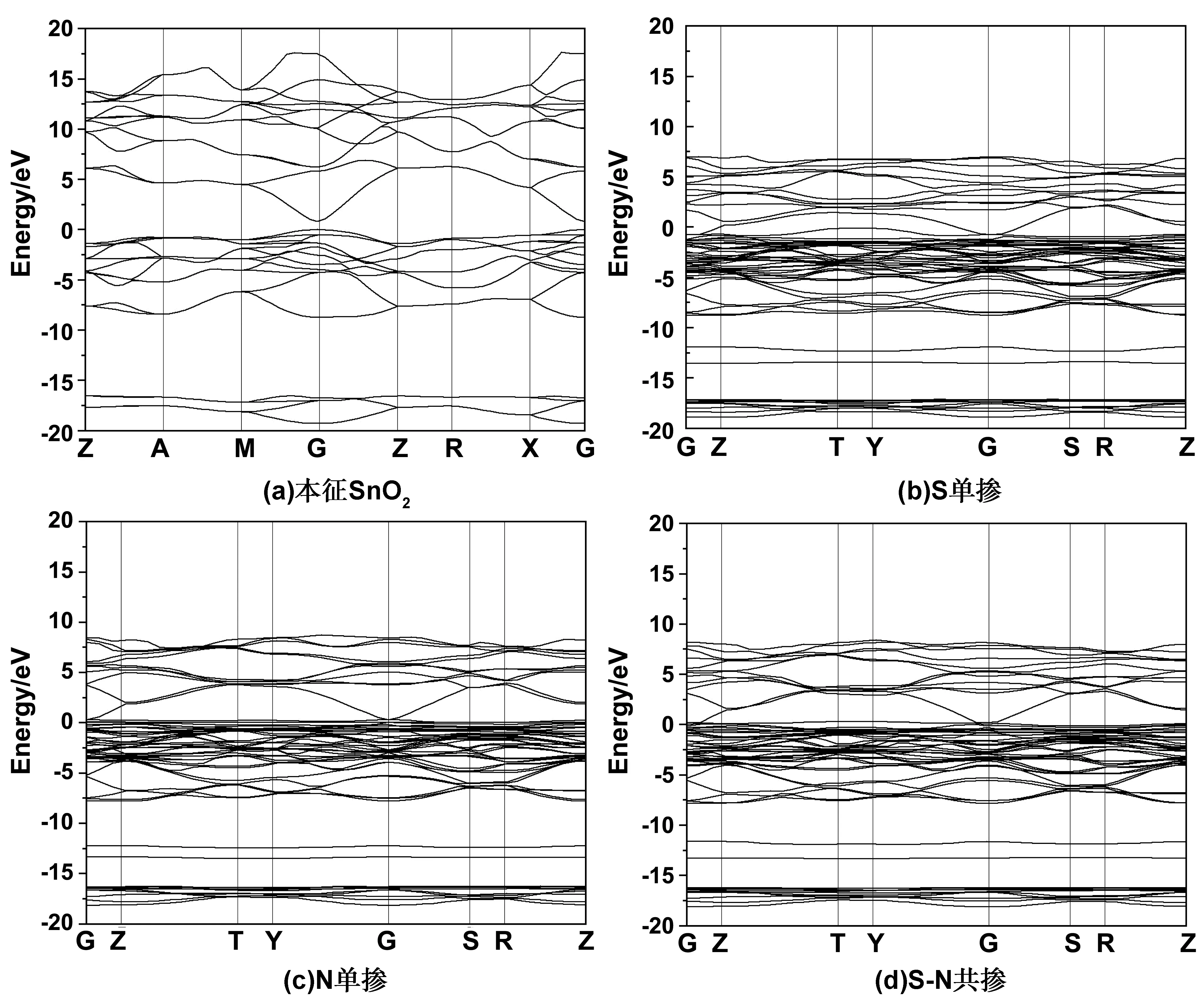
图2 SnO2掺杂能带结构
2.3 态密度
态密度主要可以反应掺杂原子和SnO2内的原子之间的成键的情况,也可以反映出导电性能。图3是本征SnO2(a)、N、S单掺(b)(c)和共掺(d)体系的SnO2的总、分态密度图,图像上的总态度反映出,掺杂后的SnO2总的态密度增加,导带向费米能级移动带隙变小,载流子浓度增加,导电性变强。本征SnO2的导带主要是由Sn原子的5s、5p态提供,范围在0~15eV[14];价带的两个峰值,由Sn原子的5s、5p轨道和O原子的2s轨道共同杂化形成的-10~0 eV的上价带,以及由Sn原子5s、5p轨道和O原子2p轨道组成的-20~-15 eV的下价带。
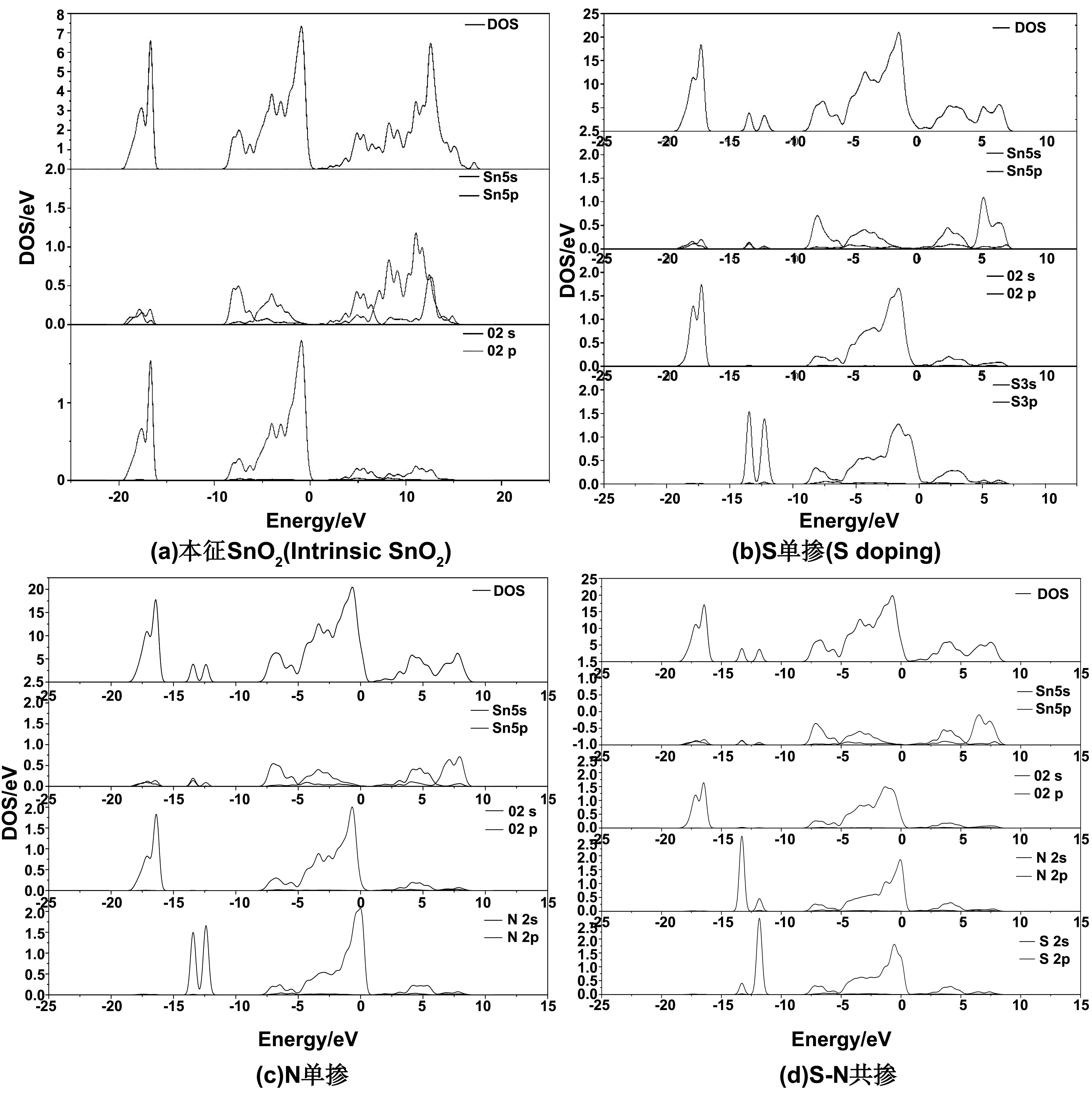
图3 掺杂各体系态密度
从图(b)的总、分态密度观察到当 S 元素单掺杂时体系导带范围便缩小至0~10 eV,导带由Sn原子的5s、5p轨道和S原子的3p轨道杂化而成;-10~0 eV这范围的态密度主要由S原子3p轨道、O原子的2p轨道以及Sn原子的5s和5p轨道杂化而成。在-12.5 eV处,S原子的3s轨道和少量的Sn原子的轨道杂化形成杂质级,使得价带的宽度增加,载流子密度增加。图(c)为N原子单掺杂体系的态密,总态密度图可知,掺杂体系的导带范围在0~10 eV之间,由Sn原子的5s、5p轨道和N原子的2p轨道杂化而成;-10~0 eV部分主要由N原子2p轨道、O原子的2p轨道以及Sn原子的5s和5p轨道杂化而成。在-12.5 eV处,N原子的2s轨道和少量的Sn原子的轨道杂化形成杂质级,使得价带的宽度增加,载流子密度增加。图(d)为N、S共掺杂体系态密度,掺杂体系的导带范围在0~10 eV之间,由 Sn原子的5s、5p轨道和N原子的2p轨道以及S原子的3p轨道杂化而成;-10~0 eV这一部分的态密度主要由N原子2p轨道、S原子的3p、O原子的2p轨道以及Sn原子的5s和5p轨道杂化而成。在-12.5eV处,S原子的3s轨道和N原子的2s轨道和少量的Sn原子5s5p共同杂化杂质级。N、S共掺杂,价带顶部态密度O2p被N2s和S3s取代,会增加更多的导电空穴,同时N和S轨道杂化后,价带顶部上移,带隙减小。电子共有化程度加强,局域性强,导电性提升。
2.4 布局分析
布局数主要体现出不同体系晶胞当中电荷转移、成键类型等情况,表2为不同掺杂体系原子电荷布局数和键重叠布局数,表内各值均取平均值。键重叠布局数大小体现原子间相互作用共价性和离子型强弱。从表中可知,本征SnO2的Sn-O成键布局数为0.535,这说明Sn原子和O原子之间共价性很强。N、S单掺杂时,掺杂原子和Sn原子发生杂化,使Sn-O键布局数减小,成键效果减弱,共价性变弱,而S和N和电负性均小于O,使得共有化程度减小,由于电负性使得Sn-S键和Sn-N键的布局数均小于Sn-O键,共价性减弱。共掺情况下,除了Sn-S键和Sn-N键布局数减小,共价性减弱,N和S之间也会成键,体系整体布局数小于单掺,进而导电性增强,强于本征和单掺体系。

表2 不同掺杂情况下各原子布局数和键重叠布局数
2.5 电导率分析
电导率δi的大小可以体现材料导电能力的强弱,电导率越大,材料的导电性能越好[15]。半导体材料导电率为:
δi=niqμi
(1)
ni表示电子浓度,q是电子电荷常量,μi电子迁移率。透过公式不难得出电导率δi和电子浓度ni还有电子迁移率μi三者之间的关系即电导率与电子浓度和电子迁移率成正比。使用origin软件对掺杂体系的态密度进行积分比便能得到电子迁移率μi。
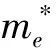
(2)
电子有效质量:
(3)
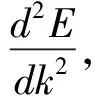
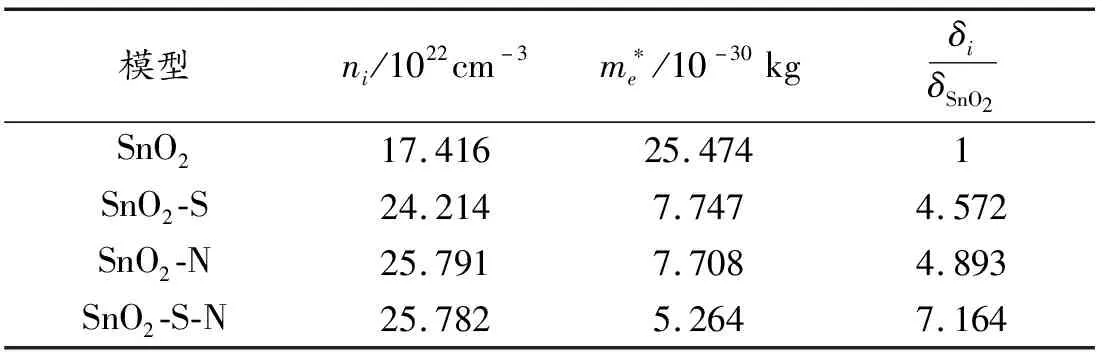
表3 SnO2掺杂前后电导率系数
可以看出,随着单掺N、S的SnO2的体系电导率小于本征SnO2体系,而共掺杂导电率最大,导电性最好。
3 结 论
本文建立了1×2×3的SnO2超晶胞结构,利用非金属原子N、S替换掉原来的O原子对SnO2进行掺杂,使用基于密度泛函理论第一性原理计算分析结构优化的本征SnO2N、S单掺杂和NS共掺杂SnO2体系的能带图、态密度和电荷分布局数。结论如下:
(1)体系掺杂后热稳定性都升高,其中共掺杂的热稳定最好;单掺和共掺后SnO2的体系带隙均变小,能带紧密导带向费米能级移动,载流子浓度增加,共掺杂效果最为明显。N、S共掺杂,价带顶部态密度O2p被N2s和S3s取代,会增加更多的导电空穴,价带顶部上移,带隙减小;
(2)分析布局电荷得知,S和N和电负性均小于O,掺杂后体系共有化程度减小,共价性减弱。共掺情况下,除了Sn-S键和Sn-N键布局数减小,共价性减弱,N和S之间也成键,体系整体布局数数减小,进而导电性增强;
(3)根据计算不同掺杂体系的电导率,掺杂前后的电导率均增大,共掺的电导率最大。
综合以上分析证明S、N共掺杂后的SnO2导电率会提升。本文理论计算分析结果可为AgSnO2电接触材料的研究提供理论参考。
