A Non-toxic and Efficient Method for Extracting DNA and RNA from Peanut
2021-03-08**
**
1. Cash Crops Research Institute, Guangxi Academy of Agricultural Sciences, Nanning 530007, China; 2. Zhongkai University of Agriculture and Engineering, Guangzhou 510225, China; 3. Sugarcane Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, Guangxi 530007, China
Abstract [Objectives] To establish a non-toxic and efficient method for extracting DNA and total RNA from peanuts and laying a solid foundation for the molecular biology study of peanuts. [Methods] Based on the principle and method of purifying nucleic acids by silica gel adsorption at high salt and low pH condition, a non-toxic and efficient method to extract peanut DNA and total RNA using cetyltrimethyl ammonium bromide (CTAB) extraction solution was designed. The quality and purity of nucleic acids were detected by agarose gel electrophoresis and nucleic acids protein analyzer, respectively. The quality of DNA was further verified by enzyme digestion and PCR amplification using molecular marker techniques. The quality of total RNA was further verified by reverse transcription (RT)-PCR of actin gene and cDNA-SCoT gene differential display technique. [Results] The agarose gel electrophoresis test showed that the peanut DNA extracted by a low-toxic and effective method is free of contamination and degradation. Through the detection by the nucleic acid protein analyzer, the DNA concentration, yield, A260/A280 and A260/A230 of 5 peanut varieties were 419.6-498.2 ng/μL, 20.98-24.91 μg/g, 1.89-1.96 and 2.03-2.28, respectively. The DNA was of high quality and can be completely digested by EcoRI restriction enzymes, and also can be used for SCoT and SRAP molecular marker technology analysis. The RNA extracted from different tissues of peanuts showed no visible DNA bands by non-denaturing agarose gel electrophoresis. The separated 28S bands were brighter than 18S. The ratio of A260/A280 and A260/A230 showed that the RNA quality was good and can be used for reverse transcription, RT-PCR of actin gene and amplification of cDNA-SCoT gene differential display technique. [Conclusions] This experiment established a low-toxic and effective method for extracting DNA and total RNA from peanuts. Compared with traditional methods, this method is more time-saving and cheaper than commercial kits. The most important point is that this method does not use toxic reagents such as phenol, chloroform and isopropanol. Thus, it is expected to be widely applied in molecular biology research.
Key words Peanut, DNA and RNA extraction, High salt and low pH, Silica-based purification
1 Introduction
Peanut (Arachis
hypogaea
L.) is one of the most important oil crops and cash crops in the world, and one of the most important sources of vegetable oil. Peanuts not only have high content of oil and protein, high nutritional value, and high economic benefits, but also can be used for biological nitrogen fixation and fertility enhancement. However, compared with model plants and other major crops, peanut molecular biology research is relatively backward. High-quality nucleic acid is an essential precondition for the study of peanut molecular biology. However, peanuts contain a large number of oils, phenols and secondary metabolites, which are not conducive to the extraction of nucleic acids. Therefore, it is very necessary to establish a low-toxic and effective extraction method for peanut nucleic acids.Based on the cleavage of guanidine thiocyanate, inactivation of ribozyme, and the feature of silica gel absorbing nucleic acids, Boomet
al
.established a method for separating nucleic acids, but the guanidine thiocyanate used in this method is highly toxic. Later, some researchers have developed a method to isolate plant DNA under high salt and low pH conditions, but this method is not effective in extracting DNA from plants rich in proteins, polysaccharides and phenolic compounds. Wanget
al
.found that the peanut DNA extracted by this method had the problem of degradation. At present, many nucleic acid extraction methods based on cetyltrimethylammonium bromide (CTAB) and sodium dodecyl sulfonate (SDS) have been reported. However, these extraction methods use high volume of toxic organic reagents, which are toxic to experiment operators and will pollute the environment. Yinet
al
.established an economical and convenient method to isolate DNA and total RNA from peanut tissue, but the method also used chloroform, a toxic reagent. Some commercial kits can alleviate these problems to a certain extent, but their cost is relatively high. Especially when the experiment requires a large amount of DNA and or total RNA, the cost will be much higher. What’s more, our research team found that when using the kit to extract RNA from different tissue parts of peanuts, the extraction effect of leaves is acceptable, but the extraction effect of roots, stems, flowers, and seeds is not good, and it is difficult to guarantee the reliability of the experimental results.Peanuts are rich in fat, protein and polysaccharides, so it is difficult to extract nucleic acids effectively. Peanut DNA extracted by traditional methods is accompanied by a certain degree of degradation; when commercial kits are used to extract peanut DNA and total RNA, the DNA extraction effect is good, but total RNA is accompanied by varying degrees of contamination. Especially when using commercial kits to extract a large amount of nucleic acid, the cost is huge. Now, the most serious is the widespread use of toxic organic reagents in traditional nucleic acid extraction methods and commercial kits.
Based on the principle and method of CTAB extraction, under high salt and low pH conditions, through purification of nucleic acid by silica gel adsorption, we established a method for extracting DNA from peanut leaves and total RNA from different tissues of peanuts. The yield and purity of the extracted nucleic acid can fully meet the needs of downstream molecular experiments. This method is expected to provide a certain degree of technical support for peanut molecular biology research.
2 Materials and methods
2.1 Materials
We collected the roots, stems, leaves and flowers of 5 peanut cultivars (Zhongkaihua 99, Guihua 836, Hanghua, Shanyou 523 and Fuhua 4).2.2 Reagents and consumables
Reagents used in this experiment included extraction buffer[100 mM Tris-HCl (pH 8.0), 25 mM EDTA, 1.5 M NaCl, 2.5% CTAB, and 2.0% PVP], high salt and low pH solution[5.0 M NaCl (pH 5.5)], TE buffer, 8 M LiCl, 75% ethanol, RNase-free water. Consumables included 1.5-2.0 mL centrifuge tubes, purification adsorption columns, RNAaseH, EcoRI, Taq DNA polymerase, and dNTPs.2.3 Methods
2.3.1
Peanut DNA extraction. Grinded the peanut leaves in liquid nitrogen. Transferred the powder into a new 2-mL centrifuge tube containing 1 mL extraction buffer (preheated at 65 ℃). Added 2 μL of RNAaseH and mixed well. Placed in a water bath at 65 ℃ for 30 min. Every 10 min, gently inverted to mix one time. Took out the centrifuge tube and centrifuged at 12 000 rpm for 10 min at room temperature. Transferred the supernatant into a new 2-mL centrifuge tube. Then, added 1 mL of 5.0 M NaCl (pH 5.5). Mixed properly (do not use vortex) and stored at -20 ℃ for 15min. Centrifuged at 13 000 rpm for 10 min at 4 ℃. Carefully transferred the supernatant to a new purification adsorption column and centrifuged at 8 000 rpm for 1 min. Repeated this step until all of the supernatant was transferred. Added 750 μL of 75% ethanol to the adsorption column, placed at room temperature for 2 min and centrifuged at 8 000 rpm for 1 min. Repeated this step 2-3 times and centrifuged at 12 000 rpm for 30-45 s. Dried and transferred the adsorption column to a new collection tube. Added 50-100 μL of TE buffer to the adsorption column for 2-3 min and centrifuged at 12 000 rpm for 1 min. Finally, used 1.2% agarose gel electrophoresis and nucleic acids protein analyzer to detect the quality of peanut genomic DNA.2.3.2
Peanut total RNA extraction. Grinded the peanut materials in liquid nitrogen. Transferred the powder to a new 2-mL centrifuge tube containing 1 mL extraction buffer quickly and mixed it immediately. Placed in a water bath at 65 ℃ for 15 min. Every 5 min, inverted to mix one time. Took out the centrifuge tube and centrifuged at 12 000 rpm for 10 min at 4 ℃. Transferred the supernatant to a new 2-mL centrifuge tube, added 500 μL of LiCl (8 M), mixed properly (do not use vortex) and stored at -20 ℃ for 1 h. Centrifuged at 12 000 rpm for 10 min at 4 ℃. Washed the precipitate by 75% ethanol twice. Added 400 μL of RNase-free water to dissolve the precipitate and then added 300 μL of 5.0 M NaCl to mix gently. Transferred the mixture to a new purification adsorption column and centrifuged at 8 000 rpm for 1 min. Added 750 μL of 75% ethanol to the adsorption column, placed on ice for 2 min and centrifuged at 8 000 rpm for 1 min. Repeated this step 2-3 times and centrifuged at 12 000 rpm for 30-45 s. Dried and transferred the adsorption column to a new collection tube. Added 40-70 μL of TE buffer to the adsorption column and placed on ice for 2-3 min and centrifuged at 12 000 rpm for 1 min. Finally, used 1.2% agarose gel electrophoresis and nucleic acids protein analyzer to detect the quality of peanut total RNA.2.3.3
Peanut DNA digestion. To verify the quality of peanut genomic DNA, 1 μg of DNA was digested with EcoRI. DNA was incubated at 37 ℃ for 3 h with 1.0U enzyme, 2 μL of digestion buffer and 17 μL of sterilized water. The digested DNA was detected on a 1.5% agarose gel.2.3.4
SCoT-PCR and SRAP-PCR. To verify the quality of peanut genomic DNA as PCR template, we used single primer termed SCoT19 derived from start codon targeted polymorphism and primer pair termed em3-me5 derived from sequence related amplified polymorphism (Table 1). The optimal PCR reaction system was: 50-100 ng genomic DNA, 10 μmol/L primers, 400 mmol/L dNTP, 1.0U rTaq polymerase and 2 μL of 10×PCR buffer. The SCoT-PCR amplification procedure was as follows: pre-denatured at 94 ℃ for 3 min; denatured at 94 ℃ for 1 min, annealed at 50 ℃ for 1 min, extended at 72 ℃ for 2 min, performed 35 cycles; and extended the last time at 72 ℃ for 5 min. SRAP-PCR amplification procedure was as follows: pre-denatured at 94 ℃ for 4 min; denatured at 94 ℃ for 1 min, annealed at 35 ℃ for 1 min, extended at 72 ℃ for 1 min, performed 5 cycles; pre-denatured at 94 ℃ for 1 min, annealed at 50 ℃ for 1 min, extended at 72 ℃ for 1 min, performed 35 cycles; extended the last time at 72 ℃ for 10 min. We separated the PCR amplification products by 1.5% agarose gel electrophoresis (1×TAE buffer), stained the gel with ethidium bromide at a concentration of 0.5 μg/mL, and then took the photo of observation results under the gel imaging system.2.3.5
cDNA synthesis, RT-PCR, and cDNA-SCoT amplification. We performed the synthesis of first-strand cDNA in accordance with the standard protocol (M-MLV, TaKaRa, Dalian, China). Oligo(dT)18 was used as the reverse transcription primer. Finally, we diluted the first strand of cDNA obtained by reverse transcription using 10-fold with water and stored at -80 ℃. To verify the suitability of cDNA as PCR template, we used primer pair termed actinF/actinR and single primer termed SCoT13 to perform RT-PCR and cDNA-SCoT (Table 1). RT-PCR amplification procedure was as follows: pre-denatured at 94 ℃ for 4 min; denatured at 94 ℃ for 30 s, annealed at 55 ℃ for 45 s, extended at 72 ℃ for 1 min, performed 30 cycles; extended the last time at 72 ℃ for 10 min. cDNA-SCoT amplification procedure was as follows: pre-denatured at 94 ℃ for 4 min; denatured at 94 ℃ for 30 s, annealed at 55 ℃ for 45 s, extended at 72 ℃ for 1 min, performed 35 cycles; extended the last time at 72 ℃ for 10 min. PCR amplification reaction system was the same as Section2.3.4
. We separated the PCR amplification products by 1.5% agarose gel electrophoresis, stained the gel with ethidium bromide at a concentration of 0.5 μg/mL, and then took the photo of observation results under the gel imaging system (UVITEC, Cambridge, UK).
Table 1 Primers used in the present study
3 Results and analysis
3.1 Peanut DNA quality detection
The extracted genomic DNA of five peanut varieties showed a main band on the agarose gel, indicating that the integrity of extracted peanut genomic DNA was good and not degraded. No foreign objects in each sample hole indicated that there was no protein contamination. No dispersive bands were found on the agarose gel indicated that there was no RNA contamination (Fig.1).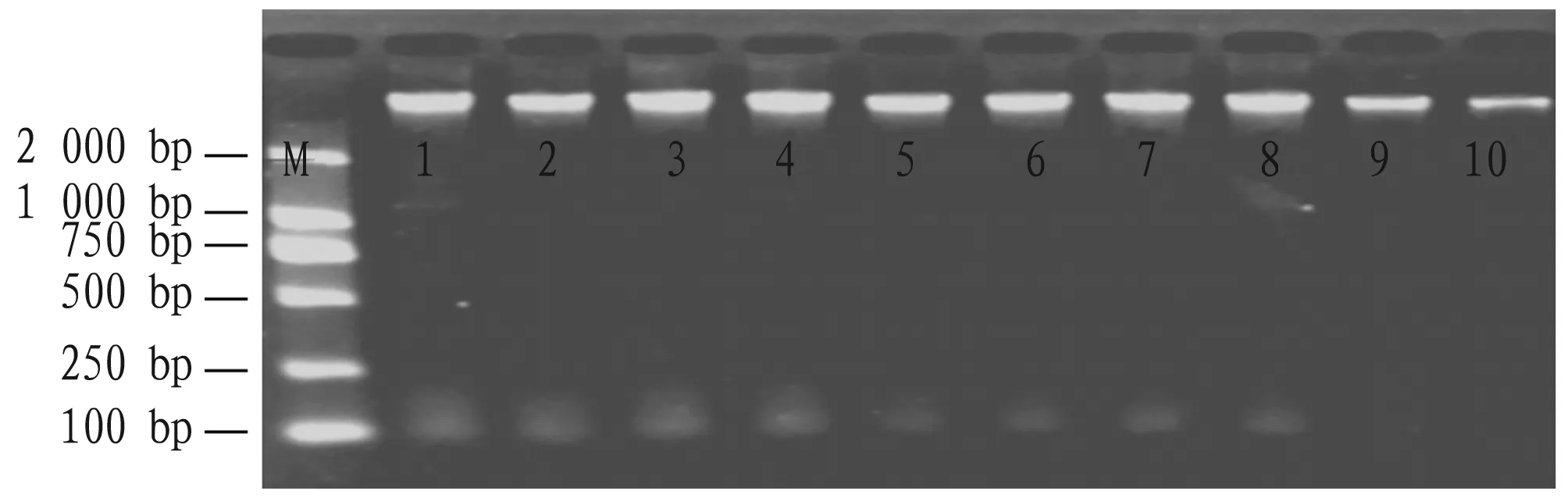
Note: M: DNA marker; 1-2: Zhongkaihua 99; 3-4: Guihua 836; 5-6: Hanghua 2; 7-8: Shanyou 523; 9-10: Fuhua 4.
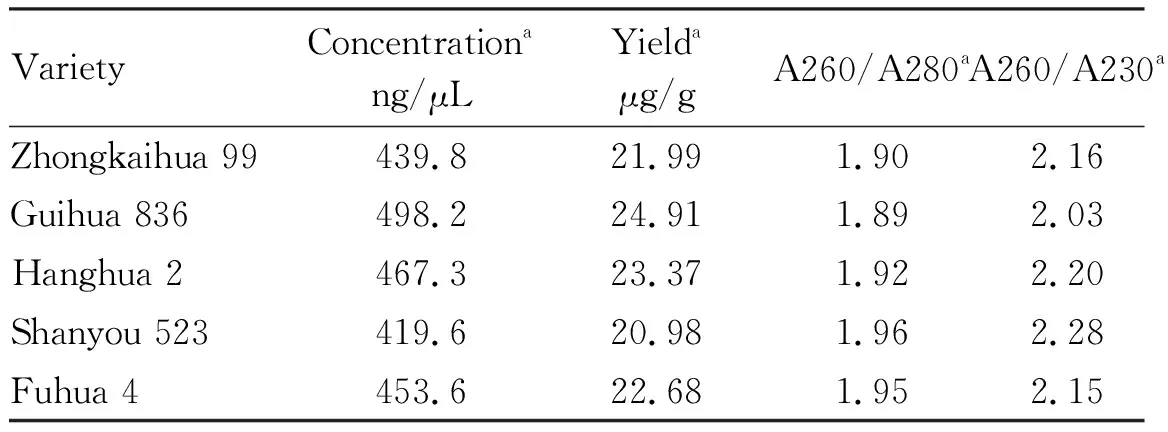
Table 2 Quality and yield of peanut genomic DNA
Note:Average of three extracts from the same tissue.
The yield of extracted genomic DNA of five peanut varieties was in the range of 20.89- 24.91 μg/g. The concentration was in the range of 419.6-498.2 ng/μL, indicating the concentration was high. The A260/A280 value was in the range of 1.89-1.96, indicating that there was no protein and phenol contamination. The A260/A230 value was in the range of 2.03-2.28, indicating that there was no polysaccharide contamination (Table 2).
3.2 Peanut DNA digestion
To further verify the quality of the extracted peanut genomic DNA, the restriction enzyme EcoRI was used to digest 1 μg of genomic DNA of the two peanut varieties. From the 1.5% agarose gel electrophoresis image, the extracted genomic DNA of the two peanut varieties can be completely digested by EcoRI (Fig.2).
Note: 1-2: Zhongkaihua 99; 3-4: Guihua 836.
3.3 SCoT-PCR and SRAP-PCR of peanut genomic DNA
To verify the suitability of peanut genomic DNA as PCR template, we used the extracted genomic DNA of four peanut varieties in PCR amplification with single primer SCoT19 and primer pair em3-me5. The agarose gel electrophoresis of SCoT-PCR and SRAP-PCR showed that the band patterns are clear, indicating that the extracted peanut genomic DNA is good enough for PCR amplification and molecular marker techniques analysis (Fig.3).
3.4 Peanut total RNA quality detection
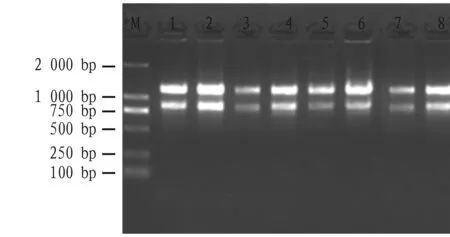
Through the non-denatured agarose gel electrophoresis, the total RNA of four tissues from four peanut varieties showed the brightness of 28S bands are higher than 18S bands, indicating that the integrity and quality of peanut total RNA were good. Few foreign objects were found in each sample hole, indicating that there was almost no protein contamination. The bands on the gel were clear without tailing, indicating that the extracted peanut total RNA had no polysaccharide contamination (Fig.4).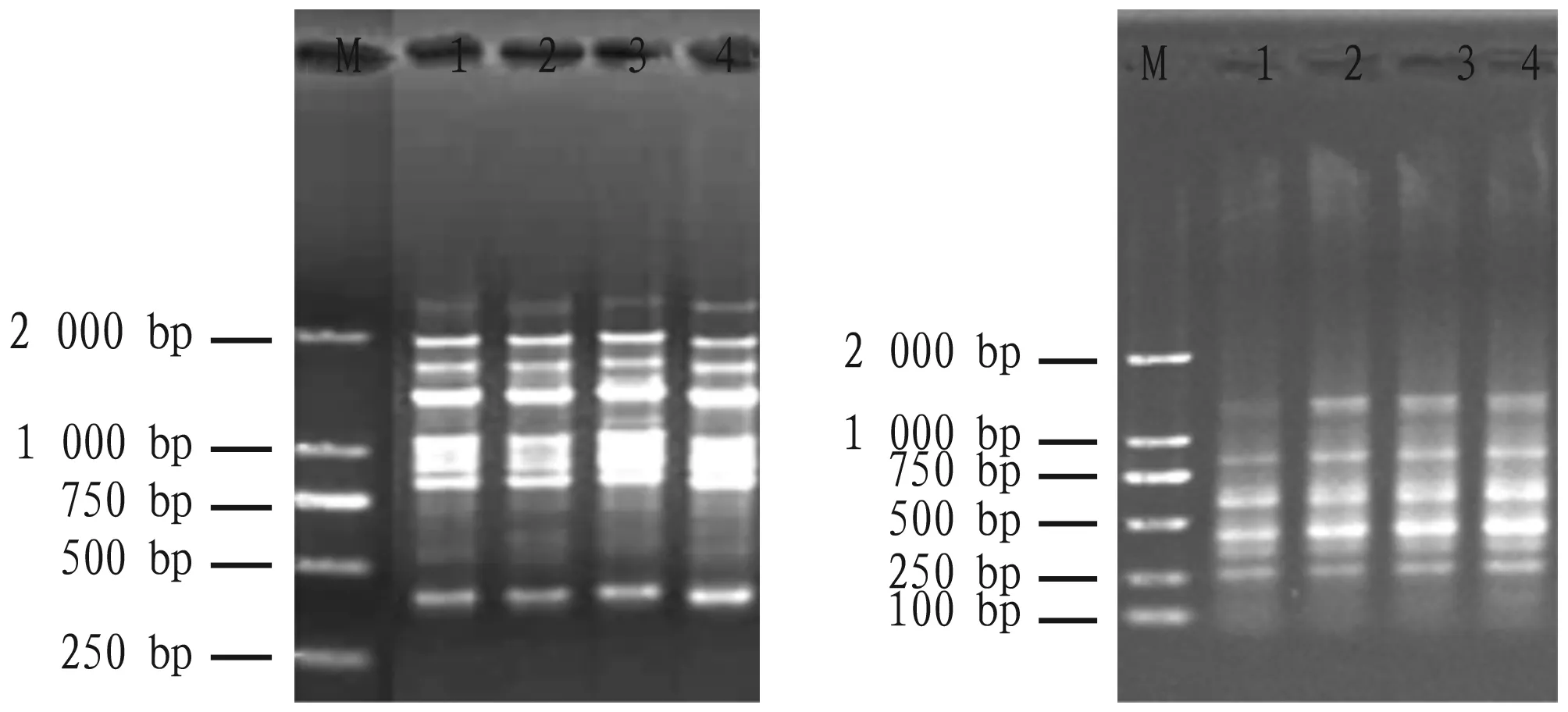
Note: M: DNA marker; 1: Zhongkaihua 99; 2: Guihua 836; 3: Hanghua 2; 4: Shanyou 523.
Note: M: DNA Marker; 1-2: Leaf; 3-4: Stem; 5-6: Flower; 7-8: Root.
We measured the concentration and yield of total RNA in flowers, leaves, stems and roots of five peanut varieties (Table 3). We found that, except for Shanyou 523, in the four tissue parts of the five peanut varieties, the concentration and yield of total RNA in leaves were the highest (Fig.5-6).
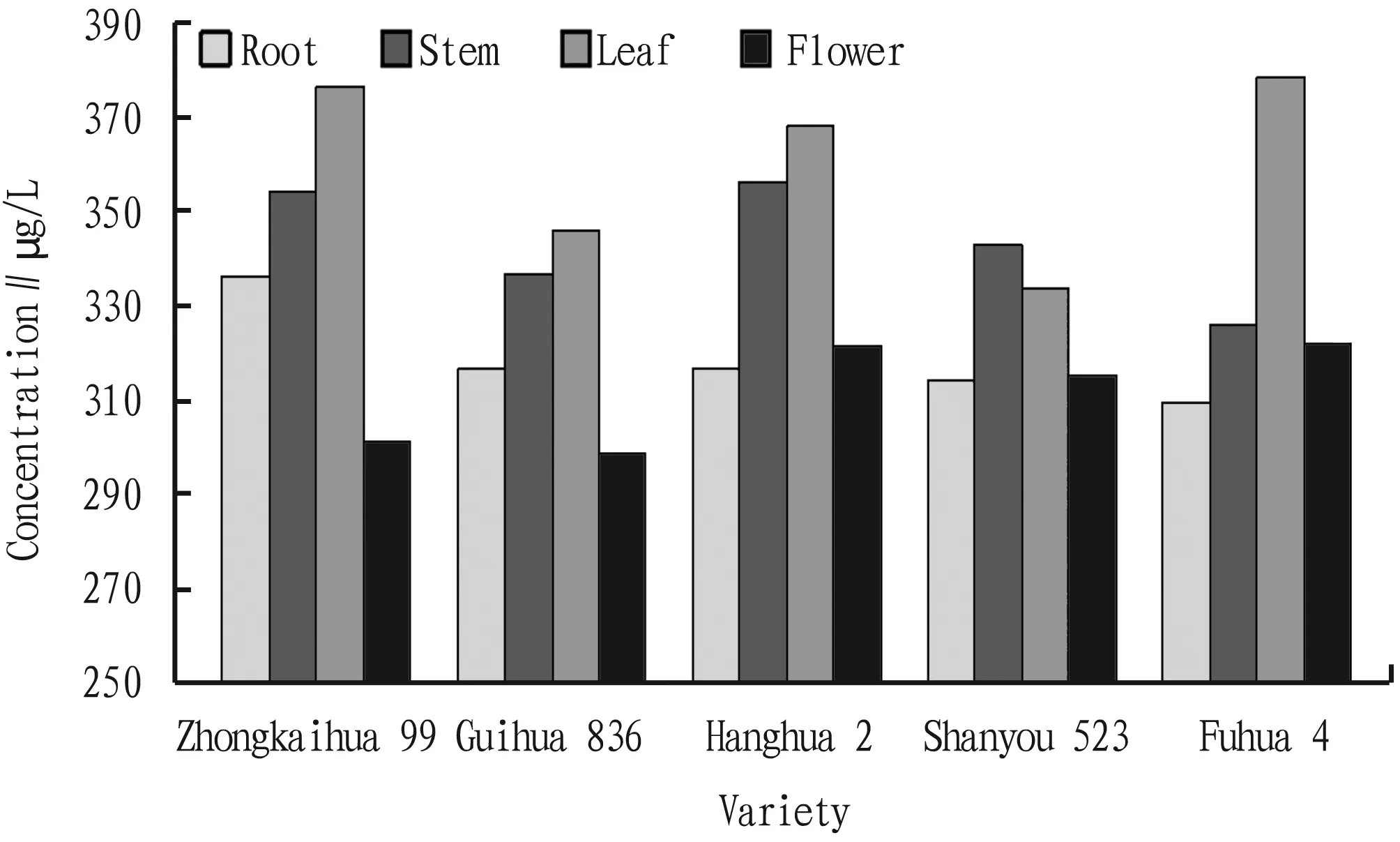
Fig.5 Concentration of total RNA from four tissues of five peanut varieties
The A260/A280 value of the total RNA in the four tissue parts of the five peanut varieties was in the range of 1.90-1.98, indicating that the total RNA of peanut had no protein and phenol contamination. The A260/A230 value of the total RNA in the four tissue parts of the five peanut varieties was in the range of 2.09-2.30, indicating that the total RNA of peanut did not have polysaccharide contamination (Table 3).

Fig.6 Yield of total RNA from four tissues of five peanut varieties
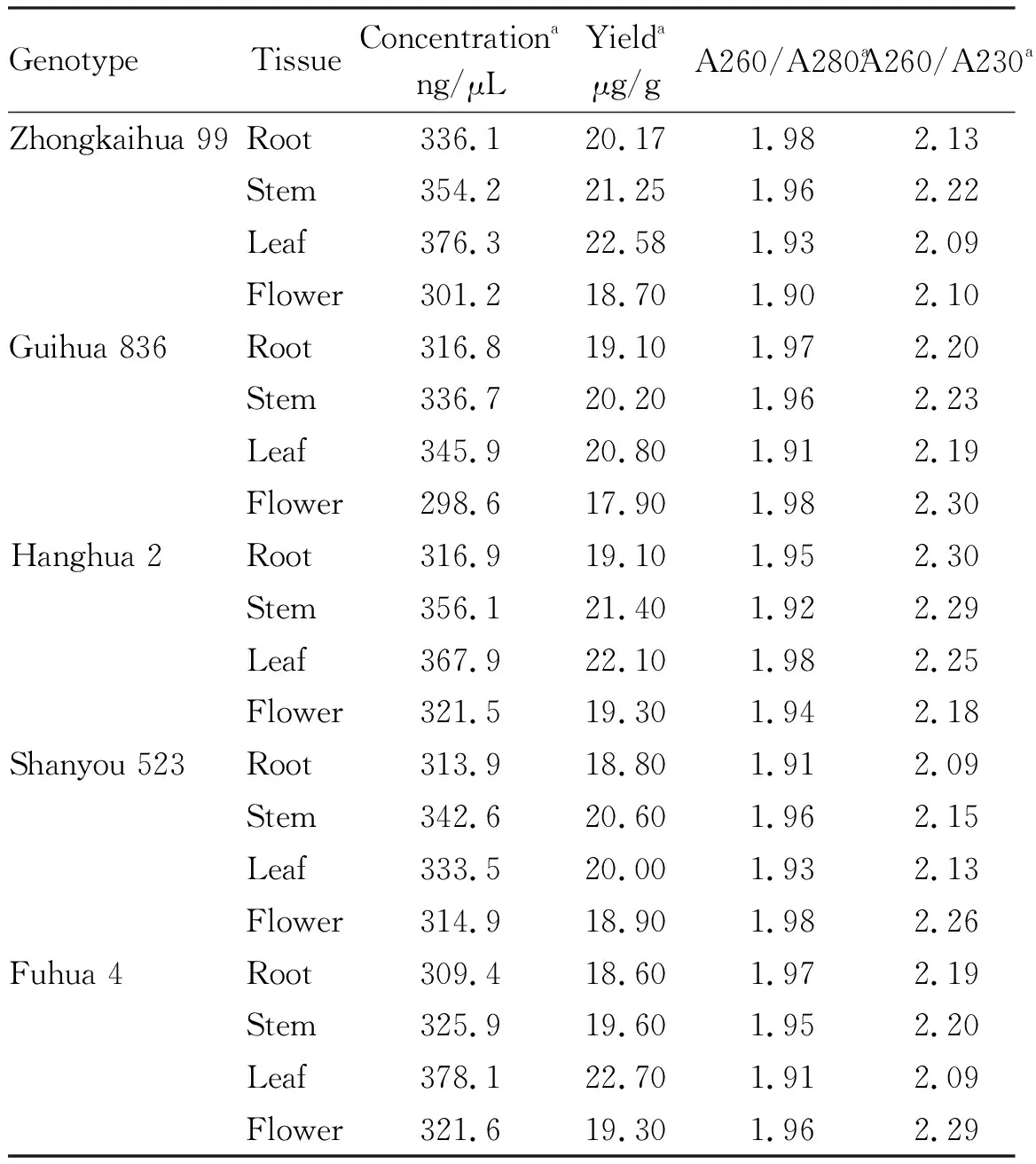
Table 3 Quality and yield of peanut total RNA
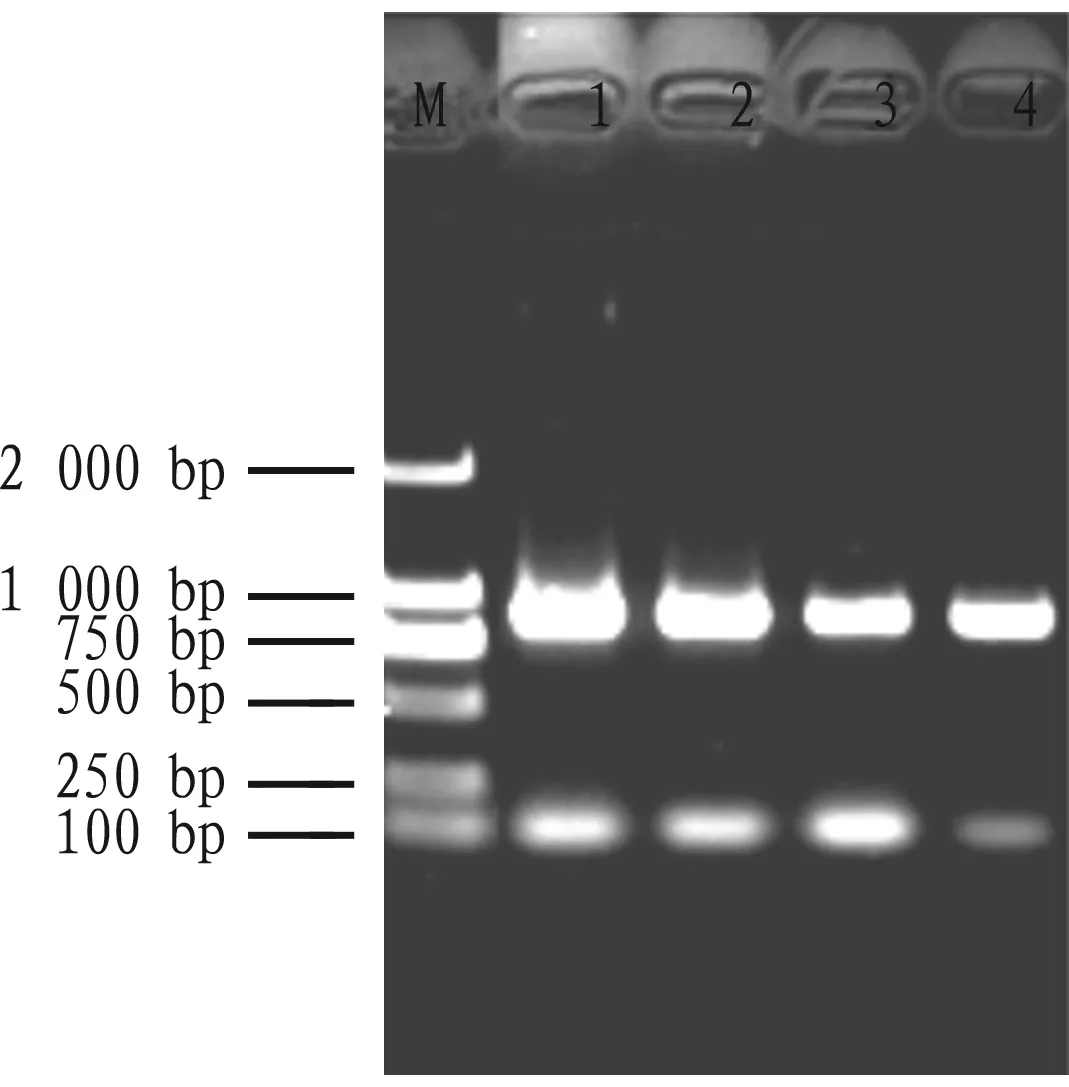
Note: M: DNA Marker; 1-2: Zhongkaihua 99; 3-4: Guihua 836.
3.5 RT-PCR and cDNA-SCoT amplification
With the cDNAs of Zhongkaihua 99 and Guihua 836 as PCR amplification templates, we performed RT-PCR and cDNA-SCoT amplification using the primer pair for amplifying peanut actin gene and the SCoT labeled single primer, respectively. As shown in Fig.7, the amplified band of the actin gene is clear. From Fig.8, it also can be seen that cDNA-SCoT can amplify multiple bands ranging from about 250 bp to 2 500 bp, indicating that the extracted RNA is of good quality and can be used for subsequent gene expression analysis.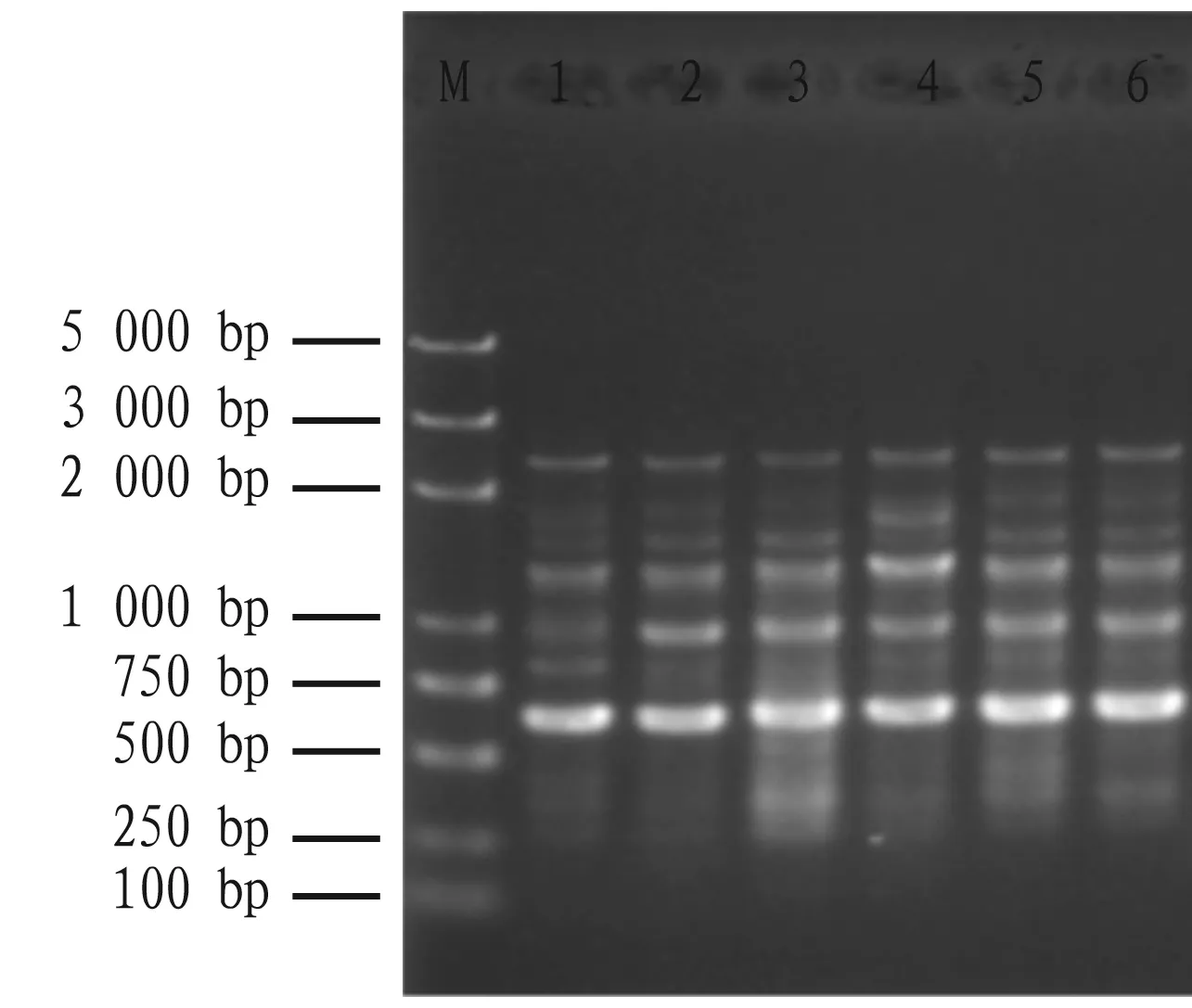
Note: M: DNA Marker; 1-2: Zhongkaihua 99; 3-4: Guihua 836; 5-6: Hanghua 2.
4 Discussions
Peanuts are rich in oils, proteins and polysaccharides. The extraction of high-quality nucleic acids requires repeated use of toxic organic reagents and takes a long time. Thus, during the DNA extraction, it is common for RNA degradation and protein and polysaccharide contamination to occur.
In recent years, commercial kits using adsorption purification columns to separate nucleic acids have been used in many laboratories. The adsorption purification column contains silica resin that can selectively bind DNA and RNA under salt conditions, but the main drawback is that researchers have never known what the extraction reagents in these commercial kits are. In this study, we developed a nucleic acid extraction method comparable to the commercial kits. The key components include sample lysis buffer, high-concentration salt solution, washing buffer and adsorption purification column. At present, there have been commercial products of adsorption purification columns. On this basis, it is only necessary to to figure out the composition of the sample lysis buffer and washing buffer. Experimental results indicate that CTAB, NaCl and ethanol are the main components of the sample lysis buffer and washing buffer.
In this study, we used 2.5% CTAB lysis buffer to separate the peanut nucleic acid. The results show that CTAB is still the best choice when a large amount of nucleic acid is necessary for research, and it can increase the yield of nucleic acid. Besides, the CTAB extraction buffer contains a high concentration of polyvinyl pyrrolidone (PVP), which can inhibit the oxidation of polyphenols. In addition, in the presence of chaotropic agents (NaI or NaClO), silica gel specifically adsorbs nucleic acids under high salt and low pH conditions; then, in a suitable alkaline environment, nucleic acids can be eluted. In the experiment, we specially used 5.0 M NaCl (pH 5.5) to mix with the supernatant and store it at -20 ℃, to provide a suitable environment for nucleic acid adsorption and precipitation of some proteins, which is also a key step of the process. We use 5.0 M NaCl (pH 5.5) instead of NaI and NaClObecause NaI and NaClOare more prone to oxidation-reduction reactions, NaCl is safe and non-toxic, and it is cheaper and easier to obtain. Furthermore, we used LiCl to precipitate RNA to avoid DNA interference. In this study, we also discussed the influence of temperature on RNA extraction and purification. The results showed that almost all steps after sample lysis can be performed at 4 ℃ to achieve the optimal effect. According to some studies, it is easier to obtain high-quality DNA from fresh young and tender leaves. The results of this study show that, except for Shanyou 523, Fig.5-6 and Table 3 indicate that among the four tissue parts of the five peanut varieties, the concentration and yield of total RNA are the highest in the leaves.
It is worth mentioning that apart from obtaining high-quality nucleic acids in this study, the entire extraction process is low-toxicity extraction, and it can greatly reduce the harm to laboratory operators. At the same time, the extracted nucleic acid is pure enough to fully meet the needs of subsequent experiments. Also, when using this method to extract nucleic acids from other plants, we also obtained good results (data not shown here).
5 Conclusions
Based on the silica gel adsorption method and the high salt and low pH method, we designed a method for extracting peanut DNA and total RNA using CTAB extract with low toxicity. The DNA extracted by this method can be used for enzyme digestion and molecular marker technology analysis, and the extracted total RNA can be used for reverse transcription, RT-PCR and cDNA-SCoT gene differential display technology. Compared with traditional methods, this method saves time; compared with commercial kits, this method costs less. Most importantly, this method does not require the use of toxic organic reagents, such as phenol, chloroform, and isopropanol. In summary, this method can be widely applied in the study of peanut molecular biology.
杂志排行

Asian Agricultural Research的其它文章
- Research on Quality Assurance System of Talent Cultivation in Higher Vocational Colleges from the Background of Enrollment Expansion
- Geographical Indication and Unique Production Technology of Zhengcheng Honeysuckle
- Thoughts on the Ways out of Breeding Industry under the Important Task of Ecological Civilization
- Development of Mountain Tourism in Central Yunnan in the Context of Rural Revitalization Strategy
- Exploration of Chinese Television Historical Comedy Drama Li Wei the Magistrate from the Perspective of Management
- Comparative Analysis of Nutritional Components in Muscles between Wild and Cultured Masu Salmon Oncorhynchus masou