Effect of Drought Stress on Proteome of Maize Grain during Grain Filling
2021-03-05
Maize Research Institute, Guangxi Academy of Agricultural Sciences, Nanning 530007, China
Abstract Based on isobaric tags for relative and absolute quantification (iTRAQ) technology, the proteome of grains of a maize cultivar Huangzao 4 under drought stress at grain filling stage was analyzed. The results show that under drought stress, 438 proteins were differentially expressed in the maize grains during grain filling. Among them, 200 were up-regulated and 238 were down-regulated. The gene ontology (GO) analysis shows that the biological processes in which differential proteins are more involved are cellular processes, metabolic processes and single biological processes; proteins in the cell component category are mainly distributed in cells, cell parts and organelles; and the proteins the molecular function category mainly possess catalytic activity and binding function. Differentially expressed proteins classified by COG are mainly involved in protein post-translational modification and transport, molecular chaperones, general functional genes, translation, ribosomal structure, biosynthesis, energy production and transformation, carbohydrate transport and metabolism, amino acid transport and metabolism, etc. The subcellular structure of the differentially expressed proteins is mainly located in the cell chloroplast and cytosol. The proportions are 35.01% and 30.21% respectively. KEGG metabolic pathway enrichment analysis shows that the differentially expressed proteins are mostly involved in antibiotic biosynthesis, microbial metabolism in different environments, and endoplasmic reticulum protein processing; the metabolic pathways with higher enrichment are the carbon fixation pathway and estrogen signaling pathway of prokaryotes; and the higher enrichment and greater significance are in the tricarboxylic acid cycle, carbon fixation of photosynthetic organisms and proteasome. The results of this study preliminarily reveal the adaptive mechanism of maize grains in response to drought stress during grain filling, providing a theoretical reference for maize drought-resistant molecular breeding.
Key words Isobaric tags for relative and absolute quantification, Grain filling stage, Maize kernel, Drought stress, Proteomics
1 Introduction
Maize (Zea
mays
L.) is the staple food crop in China and also the world. With China’s supply-side structural reform in agriculture, the planting area of maize in China has begun to decrease. In order to maintain the stability of total maize output and to guarantee the security of food supply, it is needed to further tap the yield capacity of maize. The increase in maize yield is affected by various factors. Among them, drought is the first element that restricts the development of production and the increase in yield of maize. Drought often occurs during filling stage. This period is a critical period in the growth and development of maize and is extremely sensitive to water. Investigating the impact of drought stress on the growth and development of maize during this period will be of great significance to reveal the regulation mechanism of maize grains in response to drought stress.Proteins are the executor of life activities and directly participate in the cell composition and metabolism of plants. In recent years, proteome analysis technology has developed rapidly. Two-dimensional electrophoresis technology, isobaric tags for relative and absolute quantification (iTRAQ) technology,etc
. have become the core technologies for research on proteomics. There have been many reports on the use of proteomics to study plant response and resistance mechanisms. Zhang Fengjunet
al
.used proteomics technology to study the changes in the proteome of the leaves of Qingshu No.9 with strong drought resistance under drought stress. The identified differential proteins are closely related to physiological processes such as photosynthesis, energy metabolism and antioxidant effect. Using two-dimensional electrophoresis and matrix-assisted laser desorption time-of-flight mass spectrometry, Wang Xiaxiaet
al
.analyzed the differences in the proteome of oat seeds after soaked at low temperature for 25 and 128 h and at normal temperature for 25 h, and found four proteins that may be related to low temperature stress during germination. The research on maize proteomics focuses on different tissues, such as root, stem, leaf, grainand filament, and abiotic stresses, including drought, high temperature, saltand flooding.Research on proteomics of maize grain during grain filling period has been carried out in China, and many proteins related to grain development have been identified. However, there are few reports on the law of the changes in the expression of grain development-related proteins under stresses (such as drought). The expression characteristics of drought response-related proteins in the grains of maize under drought stress at grain filling stage are still unclear. Therefore, in this study, the changes in protein expression in the gains of maize under drought stress during grain filling period were explored using the iTRAQ proteomics method, with a view to revealing the molecular mechanism of maize grains in response to drought stress during the filling stage and providing reference for the breeding of drought-tolerant maize varieties.
2 Materials and methods
2.1
Experimental
materials
and
design
The experiment was carried out in the rain shelter of Guangxi Academy of Agricultural Sciences from August to November, 2017. The maize cultivar Huangzao 4 was used as the experimental material. Two treatments were designed: normal water supply (CK) and drought stress (T). Pot planting was adopted for sowing. After sowing, 12 g of Jinzhengda controlled-release fertilizer was applied to each pot at a time. During the silking period, plants with uniform growth were selected and bagged on the same day. Pollination was performed at the same time after silking up to 5 cm. In the normal water supply treatment, the soil moisture content was maintained at 75% of the field maximum water holding capacity during the entire growth period of maize. In the drought stress treatment, water supply was cut off after pollination for 30 d. Through water control, the soil moisture content was maintained at 50%-60% of the field maximum water holding capacity. In the rest of the experimental period, the experimental conditions were the same in the two treatments. From the late milk-ripe stage to the early dough stage, three plants were selected randomly from each treatment. The kernels in the middle of the ears were collected and threshed. A total of 100 grains were collected for each treatment, quickly put into liquid nitrogen, and stored in a refrigerator at -80 ℃ for later use. Three replicates were arranged for each treatment and subjected to molecular biology research of iTRAQ proteomics.2.2
Proteome
analysis
Protein extraction, iTRAQ labeling and related bioinformatics analysis were completed by the Shanghai Mouhe Biotechnology Co., Ltd. The brief method is as follows.2.2.1
Protein extraction. A certain amount (0.5 g) of maize grains was ground thoroughly in liquid nitrogen, added with appropriate amount of lysis buffer (1∶50,W
∶V
) composed of urea (8 M), EDTA (2 mM), DTT (10 mM) and protease inhibitor (1%), ultrasonicated for 1-2 min, and centrifuged at 13 000 g for 10 min at 4 ℃ to remove impurities. The protein supernatant collected was precipitated with 3 times the volume of cold acetone at -20 ℃ for 3 h, and centrifuged at 12 000 g for 10 min at 4 ℃. The protein precipitate was re-dissolved in solubilization buffer (8 mM urea and 100 mM TEAB). The concentration of the protein was determined using a modified Bradford protein assay kit.2.2.2
Proteolysis. A certain amount (100 μg) of each sample was reduced with 10 mM DTT at 37 ℃ for 60 min, reacted with 55 mM iodoacetamide (IAM) in the dark for 30 min at room temperature, added with appropriate amount of 100 mM TEAB to make the protein concentration below 2 M, let standard at 37 ℃ overnight, subjected to enzymolysis with trypsin (protein∶trypsin=50∶1,m
∶m
), let standard at 37 ℃ for 4 h, and subjected to enzymolysis again with trypsin (protein∶trypsin=100∶1,m
∶m
).2.2.3
Peptide labeling. After trypsin digestion, the samples were desalted by Strata X SPE solid-phase extraction column and dried in vacuum. Peptide fragments were dissolved in 20 μL of 500 mM TEAB and labeled with 8-plex iTRAQ kit according to the instruction.2.2.4
HPLC separation. The dried labeled peptide fragments were dissolved in HPLC solution A (2% ACN, pH 10) and then separated using Waters Bridge Peptide BEH-Creverse-phase high-performance liquid chromatography. Simply speaking, the peptide fragments were first separated with 2%-98% acetonitrile, then fractionated into 60 tubes at a flow rate of 0.5 mL/min in pH 10 for 88 min, and finally merged into 12 tubes for vacuum centrifugal drying. According to the instruction, the samples were desalted with ZipTip C18. After vacuum drying, the samples were stored at -20 ℃ until mass spectrometry analysis.2.2.5
Identification by mass spectrometry. Each peptide fragment was identified separately by LC-MS/MS. After gradient elution with nanoLC 1000, the peptide fragments were identified by Orbitrap Elite combined mass spectrometer/Q Exactive combined mass spectrometer. A total of 20 high-intensity precursor ions were selected, identified by secondary mass spectrometry and quantified.2.2.6
Bioinformatics analysis. The mass spectrometry data were searched in the mascot and sequest databases for protein identification and quantitative analysis. Finally, bioinformatics method was used to perform functional analysis on the identified protein. In the experiment, it is defined that when the average relative expression level of certain protein is higher than 1.5 times, it is up-regulated; and when the average relative expression level is lower than 0.67 times, it is down-regulated. The protein withP
<0.05 in statistics is defined as differentially expressed protein. Differentially expressed proteins were annotated and analyzed with Omcisbean software.3 Results and analysis
3.1
Quality
control
of
mass
spectrum
data
Fig.1 shows the error distribution of the mass spectrum data. It reflects the mass deviation distribution between the actual molecular weight and the theoretical molecular weight of peptide precursor ions. The horizontal axis represents the peptide mass deviation, and the vertical axis represents the peptide score. In general, the mass deviation of most peptides was between -0.02-0.02 Da (-20-20 ppm), and the score[-log10(p)]of most peptides was above 20 points. As shown in Fig.1, the mass spectrum performance was in a relatively stable state, and the mass spectrum data meets the requirements. Fig.2 shows the distribution of peptide lengths, reflecting the state of proteolysis. The horizontal axis represents the length of the peptides, and the vertical axis represents the number of peptides under that length. In general, the length of most peptides was 7-25. As shown in Fig.2, the enzymolysis effect of protein samples was good, which met the requirements of experimental analysis.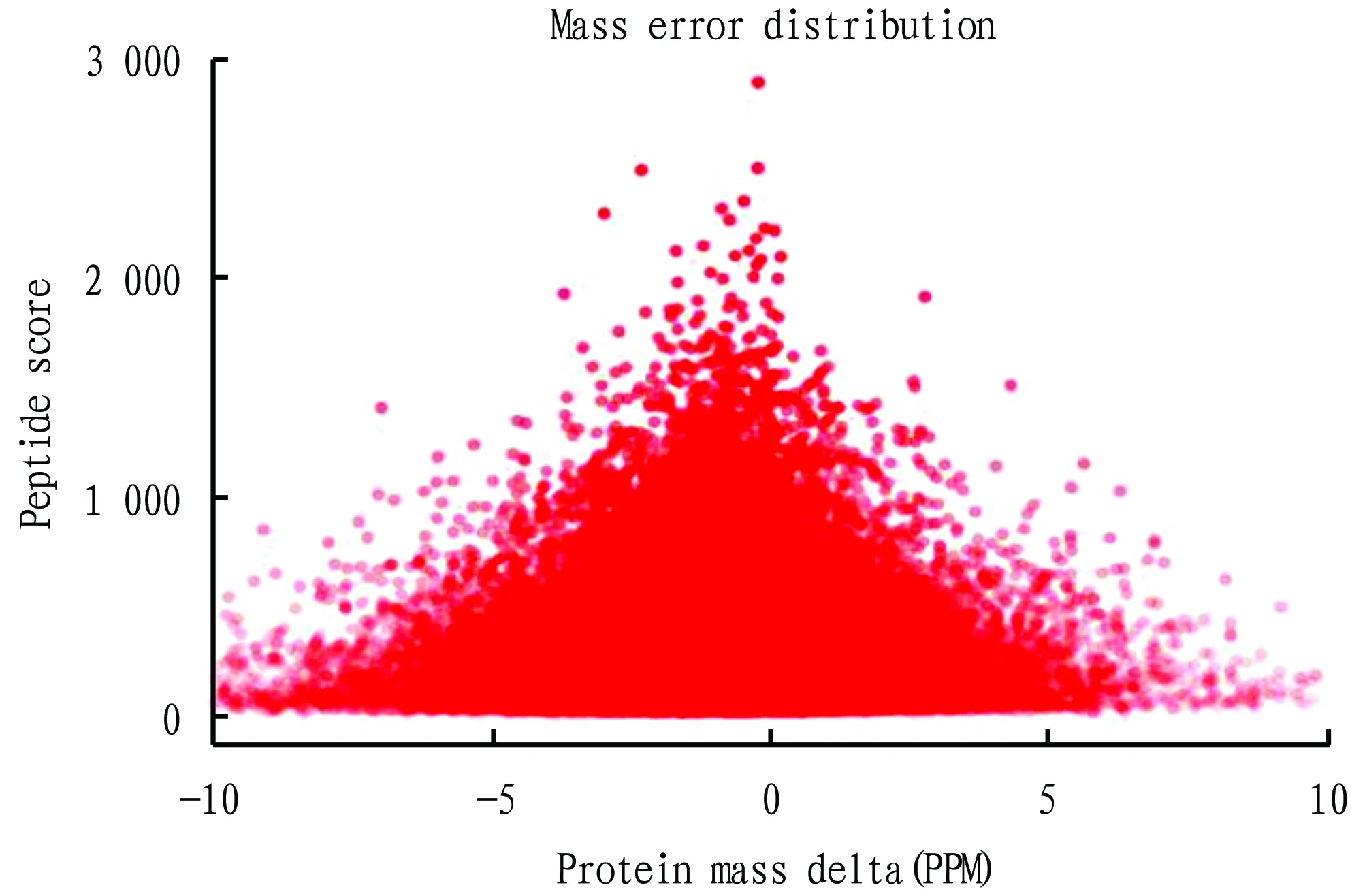
Fig.1 Distribution of mass error
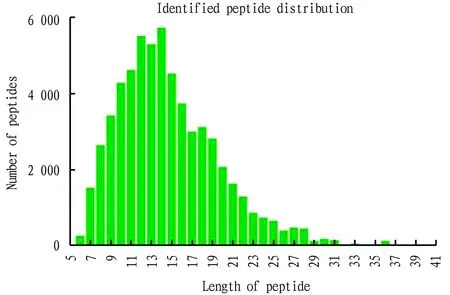
Fig.2 Distribution of peptide length
3.2
Analysis
of
differentially
expressed
proteins
in
maize
grains
under
drought
stress
during
grain
filling
The iTRAQ technology was used to analyze the proteins in the maize grains under normal water supply and drought stress during the grain filling period. A total of 4 225 proteins were successfully identified and quantified. According to the definition of differentially expressed protein, compared with normal water supply, there were 438 differentially expressed proteins in the maize grains under drought stress, of which 200 were up-regulated and 238 were down-regulated (Fig.3).3.3
GO
annotation
of
differentially
expressed
proteins
in
maize
grains
GO (Gene Ontology, http://www.geneontology.org/) is a database established by the Gene Ontology Consortium. Its purpose is to standardize the biological terms of genes and gene products in different databases, and to define and describe the functions of genes and proteins. After the maize grain proteins were annotated with GO, they were classified according to different GO levels of biological process, cellular component and molecular function. As shown in Fig.4, in the category of biological processes, proteins were widely involved in cellular process, metabolic process, single-organism process, response to stimulus, biological regulation and other processes. Among them, cellular process (106) and metabolic process (102) were the two most important biological processes, and the differentially expressed proteins involved accounted for 49.29% of the total differentially expressed proteins in biological process. In the category of cell component, proteins were mainly located in cell, cell part, organelle, macromolecular complex, organelle part and membranes. Cell (93) and cell part (90) were the two locations involving the most proteins. The differentially expressed proteins contained in them accounted for 57.19% of the total differentially expressed proteins of cell component. In the molecular function category, the proteins showed catalytic activity, binding function, structural molecule activity, antioxidant activity, molecular function regulator activity, transporter activity,etc
. Among them, catalytic activity (114) and binding function (101) were the two largest functional categories, and the differentially expressed proteins contained in them accounted for 83.66% of the total differentially expressed proteins in the category of molecular function.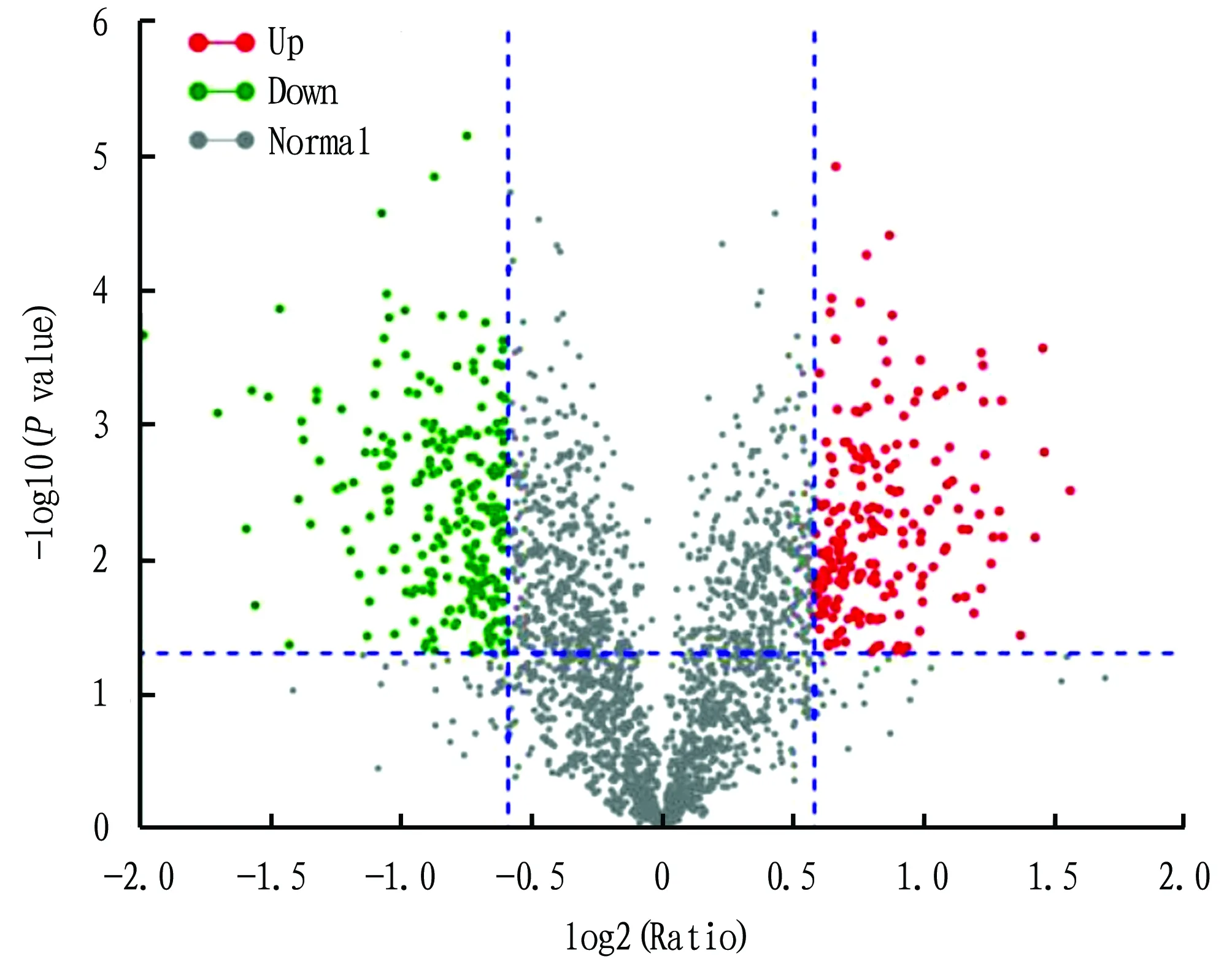
Fig.3 Distribution of differentially expressed proteins
3.4
COG
annotation
of
differentially
expressed
proteins
in
maize
grains
COG (Cluster of Orthologous Groups of proteins) is a protein database created and maintained by NCBI. It is constructed based on the phylogenetic relationship of the coded proteins of the complete genomes of bacteria, algae and eukaryotes. A certain protein sequence can be annotated into a certain COG through alignment. Each cluster of COG is composed of orthologous sequences, so that the function of the sequences can be inferred. As shown in Fig.5, the differentially expressed proteins involved in COG classification were mainly involved in post-translational modification, protein turnover and chaperones (O, 72); general function prediction (R, 40); translation, ribosomal structure and biosynthesis (Translation, ribosomal structure and biogenesis 35); energy production and conversion (C, 31); carbohydrate transport and metabolism (G, 28), amino acid transport and metabolism (E, 24),etc
.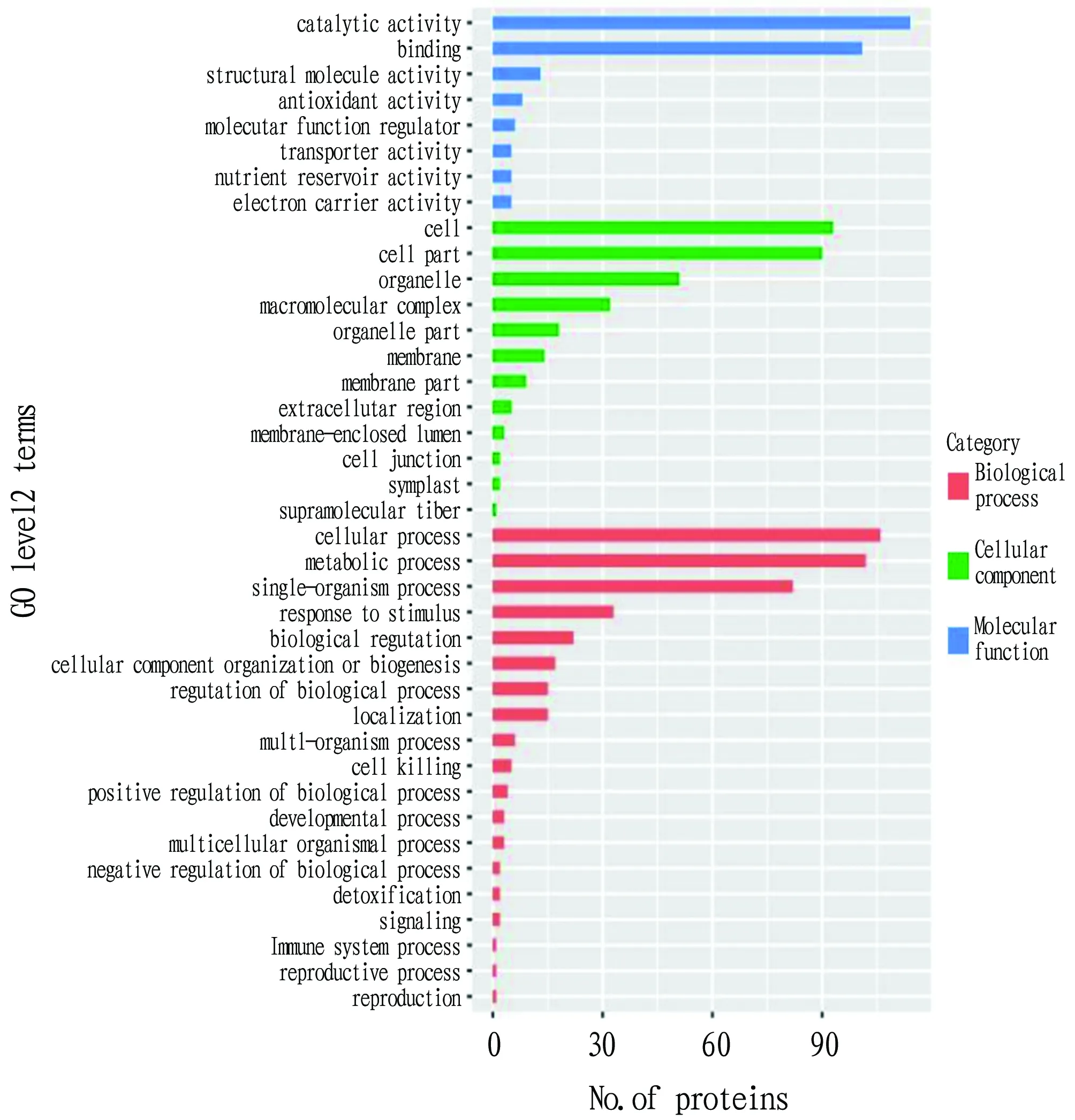
Fig.4 GO annotation of differentially expressed proteins
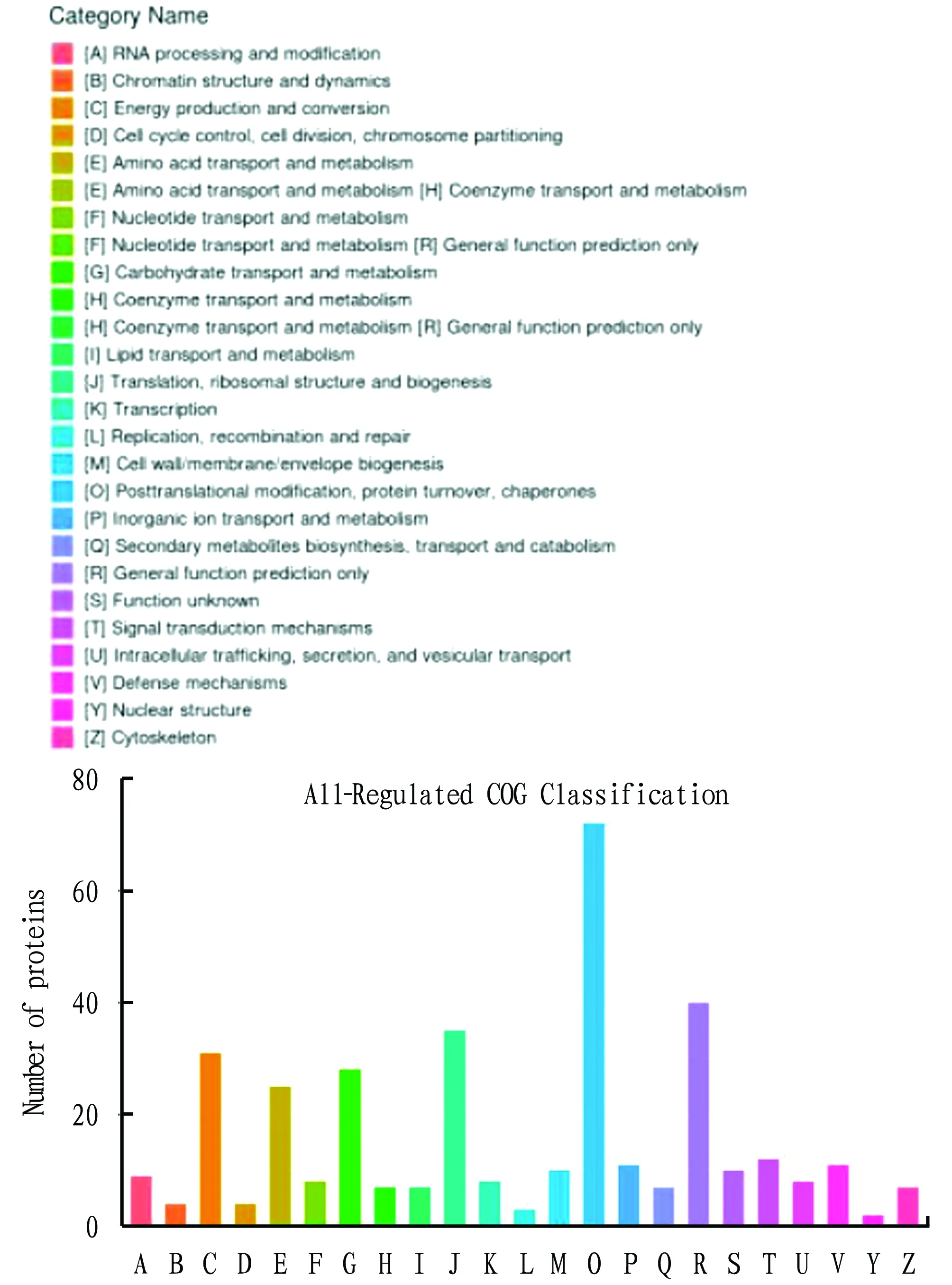
Fig.5 COG annotation of differentially expressed proteins
3.5
Subcellular
location
annotation
of
differentially
expressed
proteins
Subcellular localization analysis refers to the specific location of a certain protein or expression product in the cell. The biological function of the system is analyzed from the protein’s spatial location to provide a reference for experimental design. According to subcellular location, the differentially expressed proteins in the maize grains were classified. As shown in Fig.6, the differentially expressed proteins were mainly distributed in cell chloroplast, involving 153 proteins, accounting for 35.01%; there were 132 proteins in cytosol, accounting for 30.21%; in nucleus, 70 proteins were involved, accounting for 16.02%; and extracellular (31) and mitochondria (30) followed, accounting for 7.09% and 6.86%, respectively.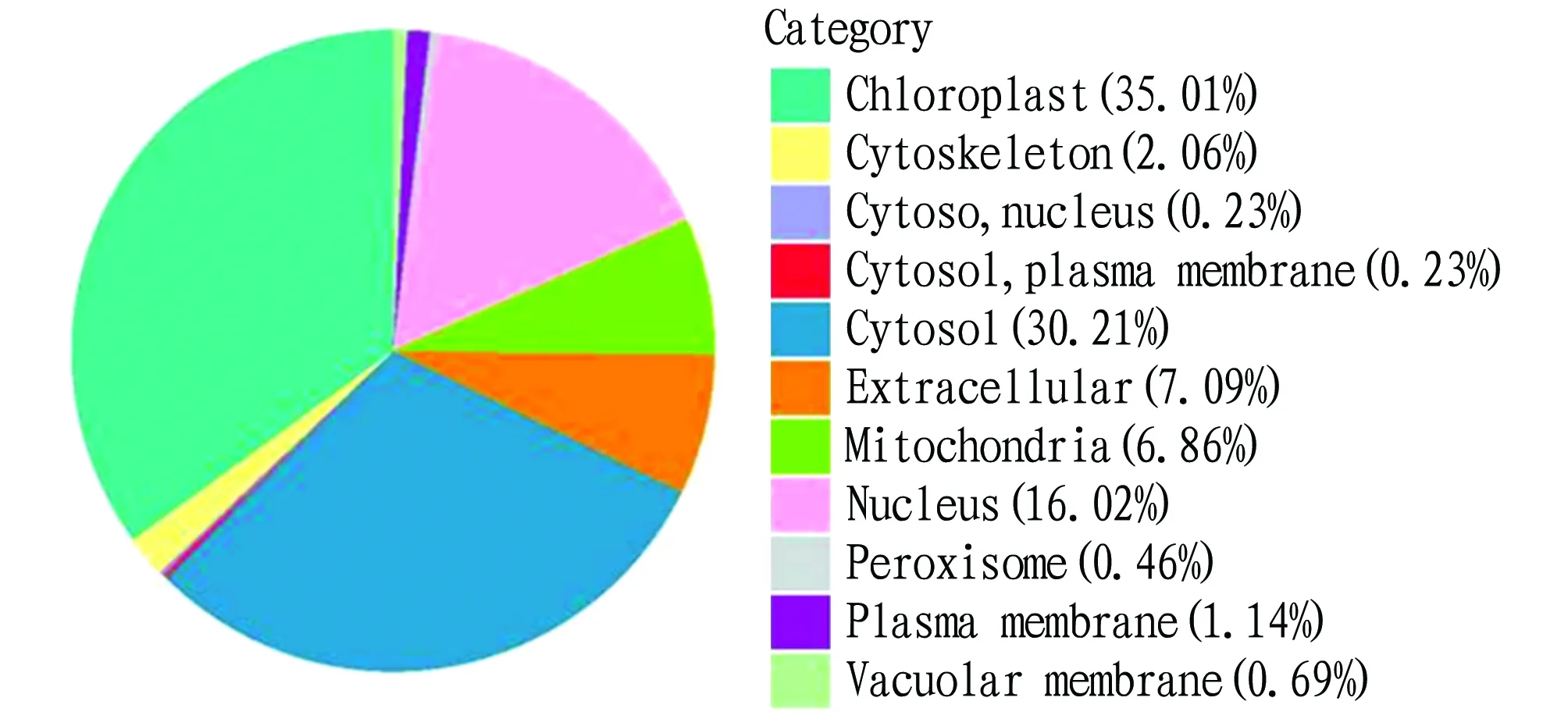
Fig.6 Subcellular location of differentially expressed proteins
3.6
Enrichment
analysis
of
KEEP
metabolic
pathways
of
differentially
expressed
proteins
in
maize
grains
KEGG provides integrated metabolic pathway query, including the metabolism of carbohydrates, nucleosides, amino acids,etc
. and the biodegradation of organic matter. It not only provides all possible metabolic pathways. Moreover, the enzymes that catalyze each step of the reaction are comprehensively annotated. Fig.7 lists the enrichment of differentially expressed proteins involved in the main metabolic pathways. Differentially expressed proteins were abundant in metabolisms such as biosynthesis of antibiotics, microbial metabolism in diverse environments, and protein processing in endoplasmic reticulum, but the enrichment significance and enrichment degree were low. The metabolic pathways with higher enrichment were the carbon fixation pathways in prokaryotes and the estrogen signaling pathway. The enrichment degree and enrichment significance in the metabolic pathways such as TCA cycle, carbon fixation in photosynthetic organisms and proteasome were high.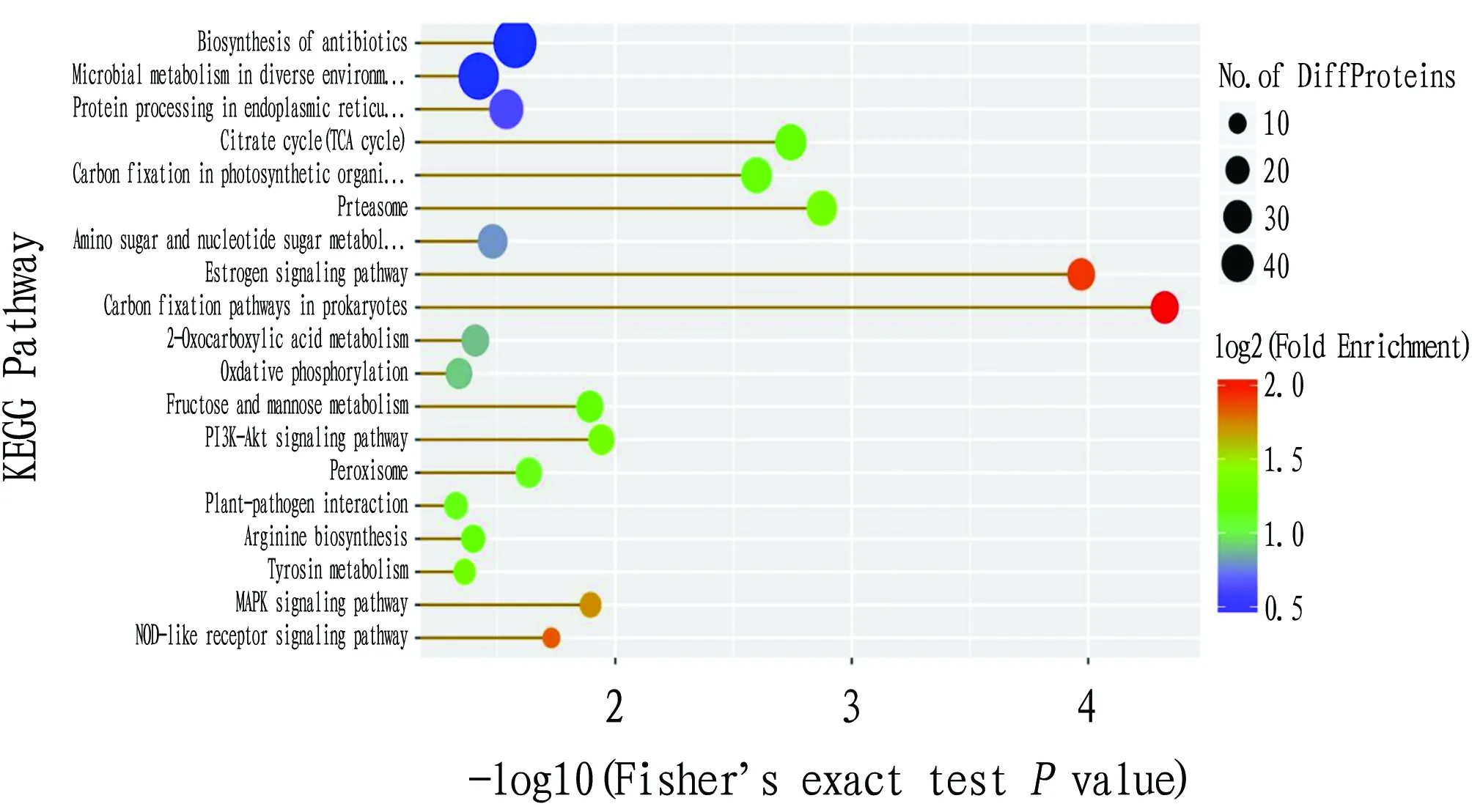
Fig.7 KEEP metabolic pathway enrichment analysis of differentially expressed proteins in maize grains
4 Discussion
iTRAQ technology is anin
vitro
polypeptide isotope labeling technology developed by Applied Biosystems in 2004. It uses 8 kinds of isotope labels to specifically label peptides. Combined with the tandem mass spectrometry, it identifies and quantifies protein samples or peptides accurately. Compared with traditional two-dimensional electrophoresis proteomics technology, it has the advantages of strong separation ability, reliable qualitative analysis results, and high sensitivity. With the continuous practice and improvement, this technology has been adopted by more and more researchers to study various regulatory mechanisms of plants. As a result, good results have been obtained. In this study, as many as 4 225 proteins were successfully isolated through iTRAQ technology, providing technical support for future research on proteome of maize grains.The results of this study show that compared with normal water supply treatment, drought stress up-regulated the expression of 200 proteins and down-regulated the expression of 238 proteins. The down-regulated proteins were more than up-regulated proteins. It indicates that compared to the increase of protein or the appearance of new protein, drought is more likely to cause the content reduction or loss of protein. GO annotation of differentially expressed proteins obtained under drought stress shows that the distribution of differentially expressed proteins in biological processes, cell components, and molecular functions are mostly the same as those of Yu Taoet
al
.in the study of maize grain development proteins. However, there are also some inconsistencies, such as cellular processes and metabolic processes in biological processes. The result of this study is that the number of differentially expressed proteins involved in cellular processes was greater than that in metabolic processes. In the study of Yu Taoet
al
, the number of development proteins involved in cellular processes was less than that involved in metabolic processes. This shows that drought stress has changed the changing trend of maize grain development protein to a certain extent. This study also annotated the differentially expressed proteins with COG. The results show that drought stress has greater effect on protein post-translational modification, transport, molecular chaperones, general functional genes, translation, ribosomal structure, biosynthesis, energy production and conversion, carbohydrate transport and metabolism and amino acid transport and metabolism. Follow-up should pay attention to in-depth research on such proteins. The results of subcellular location show that the differentially expressed proteins were more distributed in the chloroplast and cytosol. Chloroplast affects plant photosynthesis, while cytosol is related to osmotic regulation. It shows that the impact of drought stress is more reflected in the photosynthesis and osmosis of plants. These two aspects are generally considered to be directly related to the drought resistance of plants. If some key drought-resistant proteins can be found from this aspect, it will be of great help to the in-depth study of the drought resistance mechanism of maize grains during grain filling. Finally, this article carried out KEGG analysis on the differentially expressed proteins from the aspect of metabolic pathways. Both the enrichment degree and the enrichment significance were higher in the tricarboxylic acid cycle, the carbon fixation of photosynthetic organisms, and the proteasome. The tricarboxylic acid cycle is the final metabolic pathway of the three major nutrients (carbohydrates, lipids, and amino acids). The carbon fixation of photosynthetic organisms is the process of absorbing inorganic carbon and converting it into organic matter. The proteasome directly affects the turnover of certain proteins, including misfolded proteins and many proteins that play an important role in life activities. Drought stress has a great impact on important metabolic processes in these plants.In addition, this study also found some proteins that have been confirmed by previous studies to be related to drought resistance, such as superoxide dismutase and peroxidase. These two enzymes belong to the antioxidant enzyme system. Antioxidant enzymes can scavenge reactive oxygen species such as peroxides and superoxide free radicals, maintain the balance of redox and protect from oxidative damage. Their up-regulated expression is beneficial to improve the drought tolerance of maize grains. In addition, abscisic acid response protein was also identified. It is a protein induced by ABA. Under drought and water shortage conditions, it will cause a significant increase in endogenous ABA level in plants. ABA is believed to play a role in regulating cell responses in stresses. The up-regulated expression of these proteins enhances the drought tolerance of maize grains during grain filling.
Many specific proteins related to drought stress were found in this study, but they are not listed. For some key drought resistance proteins, RT-PCR or qRT-PCR was not used for preliminary verification. Therefore, follow-up experiments should further analyze the information on differentially expressed proteins based on the summary of this research, determine the key drought stress response factors, clone drought-related genes, and explain the function and mechanism of gene through genetic modification technology combined with physiological and biochemical analysis, thereby providing more theoretical basis for the breeding of drought-resistant maize varieties.
杂志排行

Asian Agricultural Research的其它文章
- Transformation and Upgrading and Countermeasures of Agricultural Industry in Xixia County, Henan Province
- Development of Ecological Agriculture in Xiantao City
- Molecular Cloning, Bioinformatics Analysis and Transcriptional Expression of Virulence-related Gene (exsA) of Vibrio alginolyticus
- Impact of Different Contour Hedgerows on Runoff, Nutrient and Soil on Sloping Farmland in Danjiangkou Reservoir Region of China