新型含有1,4-苯并二噁烷骨架化合物的设计合成与ROCK2抑制活性研究
2021-03-04杜国新张东蕾
杜国新,张东蕾
(1. 辽宁何氏医学院药学院, 辽宁 沈阳 110163; 2. 沈阳眼产业技术研究院有限公司, 辽宁 沈阳 110163)
青光眼是一组引起视神经损伤和视力丧失的眼病,以视网膜神经节细胞进行性变性为特征。几种不同类型的青光眼都有视网膜神经节细胞的死亡,神经元的细胞体在视网膜内部,它们的长轴突形成视神经,这些神经元的退化导致一系列形态改变,其中最显著的是视盘的“凹陷”(视神经损伤),视力永久丧失[1]。青光眼影响着全世界7 000 多万人,其中10%为双侧失明,是引起世界上不可逆失明的主要原因[2]。在青光眼的几种亚型中,原发性开角型青光眼和原发性闭角型青光眼在亚洲地区均较为常见,前者占比近2.3%后者占比近1.1%。由于亚洲人口规模较大,全球原发性开角型青光眼的50%以上以及原发性闭角型青光眼的75%以上发生在亚洲[3]。鉴于青光眼发病率与高龄的正相关关系,随着中国老龄化的到来,青光眼预计成为中国老年眼科疾病亟待解决的重大问题[4]。
青光眼的危险因素包括眼压升高、家族病史和高血压等。目前,青光眼的药理学治疗主要是通过减少房水的产生或改善房水的流出降低升高的眼压,降低眼压是治疗青光眼为数不多的方法之一[5]。在人眼中,有60%~80%房水经由前房角的小梁网-施莱姆式管通道流出眼外,另有约15%房水经由葡萄膜巩膜通道流出眼外[6]。但是目前临床上常见的治疗青光眼的药物,例如,β受体阻滞剂、碳酸酐酶抑制剂和前列腺素F2α类似物,它们均是通过增强葡萄膜巩膜通道的房水流出来降低眼压[7]。从现有的研究成果来看,开发一种新型的经由小梁网-施莱姆式管通道增加房水外流的药物,是对现有抗青光眼药物不同作用机制的有利补充。
ROCK 是一种丝氨酸/苏氨酸激酶,是Rho GTPase 的重要下游效应因子,在调节平滑肌组织的收缩张力中起着关键作用[8]。ROCK 有两种亚型(ROCK1 和ROCK2),两者都在人类小梁网和睫状体肌细胞中表达。多项实验证据表明,使用选择性抑制剂调节房水流出通道中的ROCK 活性,对青光眼患者眼压升高的治疗有非常显著的好处,这些抑制剂可以增加房水通过小梁网的引流,导致眼压下降[9]。截至2020年7月,目前全世界已经有两款ROCK 抑制剂治疗青光眼的药物成功上市[10]。一个是于2014年在日本上市的Ripasudil (图1),其ROCK1 和ROCK2 的IC50分别为51 nM 和19 nM;另一个是于2017年在美国上市的Netarsudil (图1),其ROCK2 的IC50为10 nM。通过查阅相关专利文献,发现化合物II (图1)报道有一定的ROCK2 活性,其IC50为10 μM[11]。
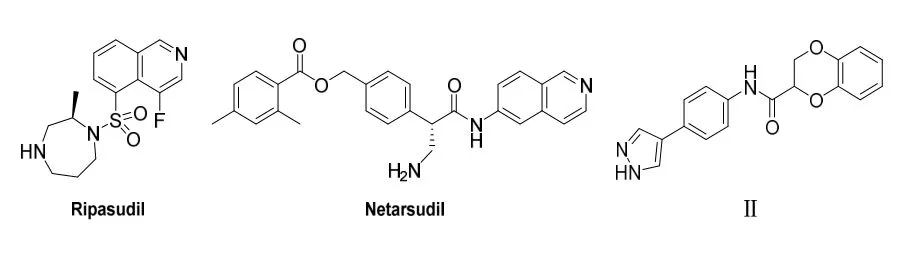
图1 已上市ROCK 抑制剂药物和化合物Ⅱ的结构
为了开发具有中国自主知识产权的,能够治疗青光眼的创新药,本论文试图找到具有全新骨架药效团的新化合物,具有一定的体外ROCK2 抑制剂活性,即可满足自主知识产权的需求,同时又能有力抵抗专利药知识产权的争夺战,为进一步开发我国治疗青光眼的创新药打下基础。
1 结果与讨论
1.1 目标化合物的设计
将化合物II 的骨架进行翻转,将活性基团与原二噁烷相连,转换成与苯环相连,与此同时活性基团由Netarsudil 的类似活性基团替代(图2)。这些活性基团包括苯甲酰基、苯乙酰基、氨基甲酰基等等,另外为了考察伯胺对活性大小的意义,还设计了伯胺被甲磺酰基取代的化合物。

图2 目标化合物10a-g, 13a-c 分子设计示意图
1.2 目标化合物的合成
化合物10a-g 按照图示1 的方法进行合成。

图示1 目标化合物10a-g 的合成路线
用TIPS-OTf 对4-羟基苯乙酸甲酯(1)进行保护,得到羟基被保护的中间体2 (91.3%)[12],中间体2 与N-溴甲基邻苯二甲酰亚胺进行烷基化反应,得到中间体3 (77.0%)[13],该中间体随后在氢氧化锂的作用下水解甲酯键和亚胺环,得到中间体4 (80.0%)[14],在DMAP 和EDCI 的存在下,中间体4 与2, 3-二氢-1, 4-苯并二氧六环-6-胺发生缩合反应,得到1,4-苯并二噁烷类似物酰胺中间体5 (68.8%)[15],而环亚胺完好无损。中间体5 在水合肼的作用下得到伯胺中间体6 (84.5%),随后在Boc2O 作用下得到氨基被Boc基团保护的中间体7 (83.0%),用四丁基氟化铵去除硅基保护基团,得到N-Boc 保护的苯酚中间体8(75.5%)[16]。中间体8 分别与相应的酰氯、羧酸和卤代烃得到中间体9a-g,随后在三氟乙酸的作用下脱掉Boc 保护基[17],得到目标化合物10a-g。
化合物13a-c 按照图示2 的方法进行合成。用中间体6 与甲磺酰氯发生胺基的酰化反应得到中间体11 (75.1%),随后用四丁基氟化铵去除硅基保护基团,得到胺甲磺酰化的苯酚中间体12 (78.9%),中间体12 分别与相应的羧酸和卤代烃得到目标化合物13a-c。所得目标化合物均经过1H NMR、13C NMR、LR MS 和HR MS 进行结构鉴定确认结构正确,经过LC MS 确认质量分数均达到95%。
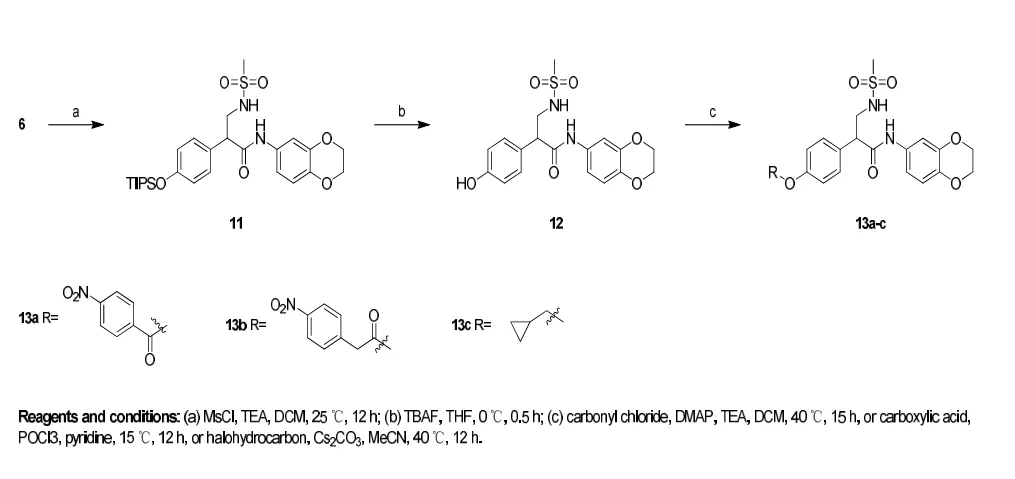
图示2 目标化合物13a-c 的合成路线
1.3 目标化合物ROCK2 抑制活性测定
用经典的 ROCK2 酶活性测试方法[18],以Netarsudil 为阳性对照品[19],测试了目标化合物10a-g和13a-c 的体外ROCK2 酶抑制剂活性,每个化合物IC50的测试结果见表1。结合化合物II 的活性结果,化合物10e 和10f 表现出了一定的体外ROCK2 酶抑制剂活性,其IC50达到了94.14 μM 和169.69 μM。从10a-g 和13a-c 的活性数据来看,药效团骨架的A环为脂肪环时活性不如芳香环;从表中可以看到13a-c 的活性不佳,说明侧链伯胺基被取代则活性下降,保留伯胺基团活性增高;在10a-g 的化合物结构中,侧链有芳香环时的活性要明显好于脂肪环的活性,且芳香环在3 位和6 位同时有取代基时,活性增加明显。
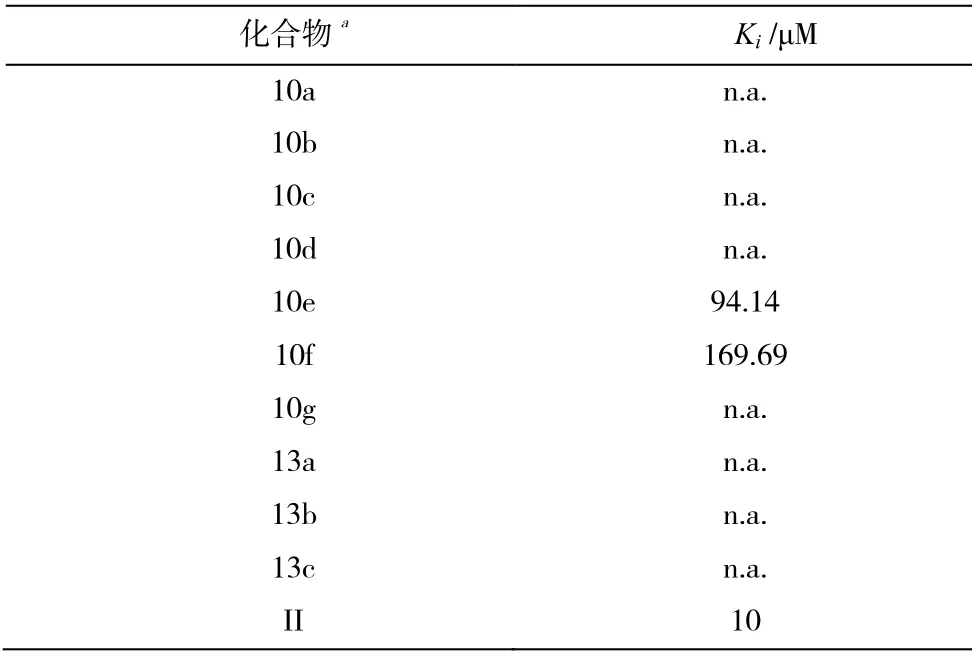
表1 化合物的ROCK2 活性结果
2 实验部分
2.1 仪器与试剂
Bruker Ascend 400、Varian S 400 MHz 型核磁共振仪, 以CDCl3、DMSO-d6、CH3OD 为溶剂, TMS 为内标; Shimadzu LC-20AB 型 HPLC; Agilent Technologies 1200 series 型 LCMS; Agilent 6210 time-of-fight LC/MS; Agela-FL-H600G 型Pre-HPLC。
2.2 实验方法
2.2.1 4-三异基硅氧基苯乙酸甲酯(2)的合成
称量4-羟基苯乙酸甲酯(1) 55.0 g 加入反应瓶中,用二氯甲烷280 mL 溶解,0 °C 搅拌下加入二甲基吡啶77.1 mL,同时加入三异丙基硅基三氟甲磺酸酯97.9 mL, 于15 °C 下搅拌反应15 h,反应液变成黄色。TLC 显示已达反应终点,加入饱和氯化铵溶液500 mL 和二氯甲烷150 mL×2 萃取,将有机层旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯=100/1 to 10/1)纯化得到中间体(2),浅黄色油状物97.5 g,收率91.3%。1H NMR (400 MHz,DMSO-d6)δ: 7.11 (d,J= 8.38 Hz, 2 H), 6.78 (d,J=8.38 Hz, 2 H), 3.62~3.53 (m, 5 H), 1.26~1.15 (m, 3 H),1.03 (d,J= 7.28 Hz, 18 H); LCMS (ESI): calcd. for C18H30O3Si [M+H]+: 323.2; found: 323.1 [M+H]+。
2.2.2 3-(N-邻苯二甲酰亚胺基)-2-(4-三异丙基硅氧基苯基)丙酸甲酯(3)的合成
称量中间体(2) 30.0 g 加入反应瓶中,用四氢呋喃300 mL 溶解,-70 °C 搅拌下加入双三甲基硅基胺基锂(1M) 97.7 mL,-70 °C 搅拌1 h,加入N-溴甲基邻苯二甲酰亚胺24.6 g,并在-70 °C 搅拌反应2 h,反应液变成黄色。TLC 显示已达反应终点,加入饱和氯化铵溶液500 mL 和乙酸乙酯200 mL×3 萃取,有机层用饱和食盐水150 mL 洗涤,将有机层旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯=100/1 to 10/1)纯化得到中间体(3),白色固体34.5 g,收率77.0%。1H NMR (400 MHz, DMSO-d6)δ:7.77(d,J =0.88 Hz, 4 H), 7.08 (d,J =8.60 Hz, 2 H), 6.72 (d,J =8.60 Hz, 2 H), 4.14~4.08 (m, 1 H), 4.04~3.99 (m, 2 H), 3.58 (s, 3 H), 3.31 (s, 2 H), 1.12 (br d,J =7.50 Hz,3 H), 0.94 (dd,J =7.28, 4.85 Hz, 18 H); LCMS (ESI):calcd. for C27H35NO5Si [M+H]+: 482.2; found: 482.2[M+H]+。
2.2.3 3-(N-(2-氨甲酰苯甲酸))-2-(4-三异丙基硅氧基苯基)丙酸(4)的合成
称量中间体(3) 124.0 g 加入反应瓶中,用四氢呋喃620 mL 溶解,同时称量氢氧化锂一水合物27.0 g用纯化水620 mL 溶解,并在20 °C 下加入到四氢呋喃溶液中,并于20 ℃搅拌反应16 h,反应液变成黄色。TLC 显示已达反应终点,加入1 N 盐酸500 mL调节pH 为4,用乙酸乙酯600 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸后得到残余物,残余物通过柱层析(二氯甲烷/四氢呋喃 = 10/1 to 2/1)纯化得到中间体(4),黄色固体100.0 g,收率80.0%。1H NMR (400 MHz, DMSO-d6)δ:13.00~ 12.32 (m, 2 H),8.43~8.33 (m, 1 H), 7.75~7.70 (m, 1 H), 7.52 ~7.43 (m,2 H), 7.20 (d,J =8.56 Hz, 3 H), 6.85 (d,J =8.56 Hz, 2 H), 3.90~3.82 (m, 1 H), 3.69~3.64 (m, 1 H), 3.54~3.44(m, 1 H), 1.30~1.19 (m, 3 H), 1.06 (d,J =7.34 Hz, 18 H); LCMS (ESI): calcd. for C26H35NO6Si [M+H]+: 486.2;found: 486.2 [M+H]+。
2.2.4 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-(1,3-邻苯二甲亚酰-2-基)-2-(4-三异丙基硅氧基苯基)丙酰胺(5)的合成
称量中间体(4) 47.0 g加入反应瓶中,用吡啶235 mL 溶解,25 °C 搅拌下加入2, 3-二氢-1, 4-苯并二氧六环-6-胺14.6 g,继续加入DMAP 11.8 g 和EDCI 36.9 g,并于25 °C 搅拌反应2 h,反应液变成红色。TLC 显示已达反应终点,将反应液旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 50/1 to 0/1)纯化得到中间体(5),黄色油状物40.0 g,收率68.8%。1H NMR (400 MHz, DMSO-d6)δ: 10.02 (s, 1H),7.80~7.71 (m, 4H), 7.17 (dd,J= 3.0, 5.6 Hz, 3H), 6.90(dd,J= 2.4, 8.8 Hz, 1H), 6.71 (dd,J= 3.4, 8.7 Hz, 3H),4.23~4.08 (m, 6H), 3.94 (dd,J= 6.5, 13.3 Hz, 1H),1.13~1.05 (m, 3H), 0.93 (dd,J= 5.5, 7.3 Hz, 18H);LCMS (ESI): calcd. for C34H40N2O6Si [M+H]+: 601.3;found: 601.3 [M+H]+。
2.2.5 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-三异丙基硅氧基苯基)丙酰胺(6)的合成
称量中间体(5) 39.0 g加入反应瓶中,用乙醇390 mL 溶解,25 °C 搅拌下加入水合肼(98%) 6.61 g,并于80 °C 搅拌反应12 h,反应液变混浊。TLC 显示已达反应终点,反应液过滤后旋蒸后得到残余物中间体(6),白色固体29.0 g,收率84.5%。1H NMR (400 MHz, MeOD)δ: 7.25 (d,J= 8.6 Hz, 2H), 7.14 (d,J=2.4 Hz, 1H), 6.91~6.83 (m, 3H), 6.72 (d,J= 8.8 Hz,1H), 4.22~4.16 (m, 4H), 3.74 (dd,J= 5.6, 8.7 Hz, 1H),3.36~3.31 (m, 1H), 2.96 (dd,J= 5.5, 12.8 Hz, 1H),1.30~1.21 (m, 3H), 1.09 (d,J= 7.3 Hz, 18H)。
2.2.6 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-(叔丁氧羰基胺基)-2-(4-三异丙基硅氧基苯基)丙酰胺(7)的合成
称量中间体(6) 28.0 g 加入反应瓶中,用二氯甲烷140 mL 溶解,25 °C 搅拌下加入Boc2O15.6 g,并于25 °C 搅拌反应2 h,反应液变混浊。TLC 显示已达反应终点,反应液过滤旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 50/1 to 0/1)纯化得到中间体(7),白色固体29.0 g,收率83.01%。1H NMR (400 MHz, CH3OD)δ: 7.26 (br d,J =8.4 Hz, 2H),7.13 (d,J =2.4 Hz, 1H), 6.92~6.80 (m, 3H), 6.74 ~6.69(m, 1H), 4.23~4.16 (m, 4H), 3.84 (dd,J =6.2, 8.6 Hz,1H), 3.61~3.52 (m, 1H), 3.43~3.34 (m, 1H), 1.40 (s,8H), 1.28~1.21 (m, 3H), 1.10 (d,J =7.3 Hz, 18H);LCMS (ESI): calcd. for C31H46N2O6Si [M+H]+: 571.3;found : 571.5[M+H]+。
2.2.7 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-(叔丁氧羰基胺基)-2-(4-羟基苯基)丙酰胺(8)的合成
称量中间体(7) 18.0 g 加入反应瓶中,用四氢呋喃90 mL 溶解,0 °C 搅拌下加入四丁基氟化铵(1M)31.5 mL,并于0 °C 搅拌反应0.5 h,反应液变成红色。TLC 显示已达反应终点,加入乙酸乙酯200 mL,用纯化水100 mL×3 萃取,有机层旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 50/1 to 0/1)纯化得到中间体(8),类白色固体10.0 g,收率75.5%。1H NMR (400 MHz, DMSO-d6)δ:9.85 (s, 1H),9.28 (s, 1H), 7.22 (d,J= 2.4 Hz, 1H), 7.11 (d,J= 8.4 Hz, 2H), 6.93 (dd,J= 2.4, 8.6 Hz, 1H), 6.84 (br t,J=5.4 Hz, 1H), 6.69 (dd,J= 8.5, 16.0 Hz, 3H), 4.16 (q,J=4.9 Hz, 4H), 3.76 (br t,J= 7.5 Hz, 1H), 3.44~3.36 (m,1H), 3.15 (td,J= 6.3, 13.0 Hz, 1H), 1.33 (s, 9H); LCMS(ESI): calcd. for C22H26N2O6[M+H]+: 415.2; found:415.1[M+H]+。
2.2.8 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(4-硝基苯甲酰氧基)苯基)丙酰胺(10a)的合成
称量中间体(8) 850 mg 加入反应瓶中,用二氯甲烷/吡啶(10/1) 20 mL 溶解,25 °C 搅拌下加入三乙胺608 mg 和DMAP25.0 mg,并加入4-硝基苯甲酰氯483 mg,并于40 °C 搅拌反应15 h,TLC 显示已达反应终点,加入饱和碳酸氢钠20 mL,用二氯甲烷40 mL 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过柱层析(石油醚/乙酸乙酯= 5/1 to 0/1)纯化得到中间体(9a),直接用于下步反应。
将得到的中间体(9a)加入反应瓶中,0 °C 搅拌下加入三氟乙酸2 mL,并于15 °C 搅拌反应3 h,LC-MS显示原料已反应完,且发现产物(10a),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10a),浅黄色固体36 mg,收率13.8%。1H NMR (400 MHz, DMSO-d6)δ:10.20 (s, 1H), 8.43 (m,J= 8.8 Hz, 2H), 8.36 (m,J= 8.8 Hz, 2H), 7.98 (br s, 3H), 7.50 (m,J= 8.8 Hz, 2H), 7.38(m,J= 8.4 Hz, 2H), 7.28 (d,J= 2.4 Hz, 1H), 6.98 (dd,J= 8.8, 2.4 Hz, 1H), 6.79 (d,J= 8.8 Hz, 1H), 4.13~4.27(m, 4H), 4.05 (dd,J= 9.2, 5.2 Hz, 1H), 3.48~3.55 (m,1H), 3.07~3.17 (m, 1H);13C NMR (101 MHz, DMSO-d6)δ: ppm 168.68 (s, 1 C), 163.59 (s, 1 C), 151.13 (s, 1 C),150.41 (s, 1 C), 143.41 (s, 1 C), 140.10 (s, 1 C), 135.41(s, 1 C), 134.86 (s, 1 C), 132.89 (s, 1 C), 131.76 (s, 1 C),129.54 (s, 1 C), 124.50 (s, 1 C), 122.67 (s, 1 C), 117.23(s, 1 C), 113.12 (s, 1 C), 109.12 (s, 1 C), 64.67 (s, 1 C),64.43 (s, 1 C), 49.58 (s, 1 C), 41.50 (s, 1 C); LCMS[M-H]+= 462.1; HR-MS calcd. for C24H21N3O7[M+H]+:464.1; found: 464.148 10 [M+H]+。
2.2.9 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(4-甲基哌嗪甲酰氧基)苯基)丙酰胺(10b)的合成
称量中间体(8) 100 mg 加入反应瓶中,用二氯甲烷3 mL 溶解,25 °C 搅拌下加入三乙胺146 mg 和DMAP2.95 mg,并加入4-甲基哌嗪甲酰氯102 mg,并于40 °C 搅拌反应15 h,TLC 显示已达反应终点,加入饱和碳酸氢钠10 mL,用二氯甲烷30 mL 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过柱层析(石油醚/乙酸乙酯= 5/1 to 0/1)纯化得到中间体(9b),直接用于下步反应。
将得到的中间体(9b)加入反应瓶中,0 °C 搅拌下加入三氟乙酸3 mL,并于15 °C 搅拌反应2 h,LC-MS 显示原料已反应完,且发现产物(10b),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10b),无色油状物36 mg,收率43.7%。1H NMR (400 MHz,DMSO-d6)δ:10.19 (s, 1H), 8.00 (br s, 3H), 7.41 (m,J=8.4 Hz, 2H), 7.26 (d,J= 2.4 Hz, 1H), 7.17 (m,J= 8.4 Hz, 2H), 6.96 (dd,J= 8.8, 2.4 Hz, 1H), 6.77 (d,J= 8.8 Hz, 1H), 4.14~4.24 (m, 4H), 4.01 (br dd,J= 9.2, 5.2 Hz,1H), 3.30~3.52 (m, 9H), 3.06 (br d,J= 9.4 Hz, 1H),2.83 (s, 3H);13C NMR (101 MHz, DMSO-d6)δ: ppm 168.73 (s, 1 C), 152.98 (s, 1 C), 150.91 (s, 1 C), 143.37(s, 1 C), 140.03 (s, 1 C), 134.57 (s, 1 C), 132.92 (s, 1 C),129.19 (s, 1 C), 122.59 (s, 1 C), 117.21 (s, 1 C), 113.07(s, 1 C), 109.06 (s, 1 C), 64.65 (s, 1 C), 64.41 (s, 1 C),52.41 (s, 1 C), 49.50 (s, 1 C), 42.85 (s, 1 C), 41.47 (s, 1 C), 40.66~40.70 (m, 1 C); LCMS [M+H]+= 441.1;HR-MS calcd. for C23H28N4O5[M+H]+: 441.2; found:441.201 84 [M+H]+。
2.2.10 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(N-乙基甲基氨基甲酰氧基)苯基)丙酰胺(10c)的合成
称量中间体(8) 150 mg 加入反应瓶中,用二氯甲烷3 mL 溶解,25 °C 搅拌下加入三乙胺110 mg 和DMAP4.42 mg,并加入N-乙基甲基氨基甲酰氯66.0 mg,并于40 °C 搅拌反应12 h,TLC 显示已达反应终点,加入饱和碳酸氢钠10 mL,用二氯甲烷30 mL萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 5/1 to 0/1)纯化得到中间体(9c),直接用于下步反应。将得到的中间体(9c)加入反应瓶中,0 °C 搅拌下加入二氯甲烷5 mL 溶解,将三氟乙酸0.5 mL 和二氯甲烷4.5 mL 的混合溶液滴加到上述溶液中,并于15 °C 搅拌反应5 h,LC-MS 显示原料已反应完,且发现产物(10c),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10c),白色固体31 mg,收率15.5%。1H NMR (400 MHz, DMSO-d6)δ:9.94~10.08 (m, 1H), 7.34(m,J= 8.4 Hz, 2H), 7.27 (d,J= 2.4 Hz, 1H), 7.06 (m,J= 8.4 Hz, 2H), 6.97 (dd,J= 8.8, 2.4 Hz, 1H), 6.75 (d,J= 8.8 Hz, 1H), 4.19 (q,J= 4.8 Hz, 4H), 3.65 (dd,J= 8.8,5.36 Hz, 1H), 3.40 (q,J= 6.8 Hz, 1H), 3.29 (q,J=6.8Hz, 1H), 3.17 (br dd,J= 12.0, 9.2 Hz, 1H), 3.00 (s,2H), 2.88 (s, 3H), 2.78 (br dd,J= 12.4, 5.2 Hz, 1H),1.01~1.20 (m, 3H);13C NMR (101 MHz, DMSO-d6)δ:ppm 171.08 (s, 1 C), 154.10 (br d,J=10.90 Hz, 1 C),150.63 (s, 1 C), 143.34 (s, 1 C), 139.69 (s, 1 C), 136.34(s, 1 C), 133.37 (s, 1 C), 128.98 (s, 1 C), 122.19 (s, 1 C),117.13 (s, 1 C), 112.86 (s, 1 C), 108.79 (s, 1 C), 64.64 (s,1 C), 64.38 (s, 1 C), 55.85 (s, 1 C), 45.97 (s, 1 C), 43.92(s, 1 C), 34.18 (br d,J=34.88 Hz, 1 C), 13.11 (br d,J=79.20 Hz, 1 C); LCMS [M+H]+= 400.1; HR-MS calcd.for C21H25N3O5[M+H]+: 400.2; found: 400.185 53[M+H]+。
2.2.11 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(3-硝基苯乙酰氧基)苯基)丙酰胺(10d)的合成
称量中间体(8) 150 mg 和3-硝基苯乙酸78.6 mg加入反应瓶中,用吡啶2 mL 溶解,0 °C 搅拌滴加三氯氧磷55.5 mg,并于15 °C 搅拌反应12 h,TLC显示已达反应终点,加入饱和碳酸氢钠2 mL,用二氯甲烷30 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到中间体(9d),直接用于下步反应。
将得到的中间体(9d)加入反应瓶中,0 °C 搅拌下加入二氯甲烷5 mL 溶解,将三氟乙酸0.5 mL 和二氯甲烷4.5 mL 的混合溶液滴加到上述溶液中,并于15 °C 搅拌反应5 h,LC-MS 显示原料已反应完,且发现产物(10d),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10d),浅黄色固体20 mg,收率9.23%。1H NMR (400 MHz, DMSO-d6)δ:10.16 (s, 1H),8.26~8.37 (m, 1H), 8.18 (br d,J= 8.4 Hz, 1H), 7.95 (br s, 3H), 7.86 (br d,J= 7.6 Hz, 1H), 7.67 (t,J= 8.0 Hz,1H), 7.43 (d,J= 8.4 Hz, 2H), 7.13~7.30 (m, 3H), 6.95(dd,J= 8.8, 2.4 Hz, 1H), 6.77 (d,J= 8.8 Hz, 1H),4.14~4.26 (m, 6 H), 4.00 (br dd,J= 8.8, 5.2 Hz, 1H),3.48 (br s, 1H), 3.00~3.15 (m, 1H);13C NMR (101 MHz,DMSO-d6)δ: ppm 169.99 (s, 1 C), 168.68 (s, 1 C),150.45 (s, 1 C), 148.24 (s, 1 C), 143.39 (s, 1 C), 140.07(s, 1 C), 137.07 (s, 1 C), 136.62 (s, 1 C), 135.02 (s, 1 C),132.87 (s, 1 C), 130.30 (s, 1 C), 129.43 (s, 1 C), 124.93(s, 1 C), 122.58 (s, 1 C), 117.21 (s, 1 C), 113.10 (s, 1 C),109.10 (s, 1 C), 64.65 (s, 1 C), 64.41 (s, 1 C), 49.52 (s, 1 C), 41.50 (s, 1 C), 40.69~40.75 (m, 1 C); LCMS [M+H]+= 478.0; HR-MS calcd. for C25H23N3O7[M+H]+: 478.1;found: 478.159 05 [M+H]+。
2.2.12 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(3-硝基-6-甲氧基苯甲氧基)苯基)丙酰胺(10e)的合成
称量中间体(8) 150 mg 加入反应瓶中,用乙腈30 mL 溶解,25 °C 搅拌下加入3-硝基-6-甲氧基苄基溴93.5 mg 和碳酸钾55.0 mg,并于40 °C 搅拌反应12 h,TLC 显示已达反应终点,加入乙酸乙酯50 mL 后过滤,滤液旋蒸得到中间体(9e),直接用于下步反应。
将得到的中间体(9e)加入反应瓶中,0 °C 搅拌下加入二氯甲烷5.0 mL 溶解,将三氟乙酸1.0 mL 和二氯甲烷4.0 mL 的混合溶液滴加到上述溶液中,并于15 °C 搅拌反应12 h,LC-MS 显示原料已反应完,且发现产物(10e),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10e),类白色固体53 mg,收率26.2%。1H NMR (400 MHz, DMSO-d6)δ:10.10 (s, 1H), 8.28 (br s, 2H), 7.93 (br s, 3H), 7.32 (br d,J= 8.8 Hz, 2H), 7.27(br d,J= 12.4 Hz, 1H), 7.07 (br d,J= 8.4 Hz, 2H),6.88~7.01 (m, 1H), 6.77 (d,J= 8.4 Hz, 1H), 5.12 (s,2H), 4.19 (br d,J= 4.0 Hz, 4H), 3.90~ 4.04 (m, 4H),3.46 (br s, 1H), 3.05 (br s, 1H);13C NMR (101 MHz,DMSO-d6)δ: ppm 169.10 (s, 1 C), 162.42 (s, 1 C),158.16 (s, 1 C), 143.35 (s, 1 C), 141.09 (s, 1 C), 139.95(s, 1 C), 133.01 (s, 1 C), 129.89 (s, 1 C), 129.50 (s, 1 C),126.63 (s, 1 C), 126.15 (s, 1 C), 124.21 (s, 1 C), 117.20(s, 1 C), 115.53 (s, 1 C), 112.99 (s, 1 C), 111.99 (s, 1 C),108.97 (s, 1 C), 64.64 (s, 1 C), 64.40 (s, 1 C), 64.32 (s, 1 C), 57.18 (s, 1 C), 49.30 (s, 1 C), 41.55 (s, 1 C); LCMS[M+H]+=480.2; HR-MS calcd. for C25H25N3O7[M+H]+:480.1; found: 480.163 90 [M+H]+。
2.2.13 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-(3-氟苯甲氧基)苯基)丙酰胺(10f)的合成
称量中间体(8) 150 mg 加入反应瓶中,用四氢呋喃10 mL溶解,25 °C 搅拌下加入3-氟苄基溴68.4 mg和碳酸铯124 mg,并于40 °C 搅拌反应12 h,TLC显示已达反应终点,加入乙酸乙酯50 mL 后过滤,滤液旋蒸得到中间体(9f),直接用于下步反应。
将得到的中间体(9f)加入反应瓶中,0 °C 搅拌下加入二氯甲烷5.0 mL 溶解,将三氟乙酸0.5 mL 和二氯甲烷4.5 mL 的混合溶液滴加到上述溶液中,并于15 °C 搅拌反应5 h,LC-MS 显示原料已反应完,且发现产物(10f),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10f),白色固体120 mg,收率57.6%。1H NMR (400 MHz, DMSO-d6)δ:9.94 (br d,J= 10.0 Hz,1H), 7.39~7.47 (m, 1H), 7.22~7.32 (m, 5H), 7.07~7.18(m, 1H), 6.97 (br d,J= 8.8 Hz, 3H), 6.75 (d,J= 8.8 Hz,1H), 5.10 (s, 2H), 4.19 (q,J= 4.8 Hz, 4H), 3.58 (br dd,J= 8.8, 5.50 Hz, 1H), 3.14 (br dd,J= 12.4, 9.2 Hz, 1H),2.68 (br s, 3H);13C NMR (101 MHz, DMSO-d6)δ: ppm 171.41 (s, 1 C), 163.88 (s, 1 C), 161.45 (s, 1 C), 157.54(s, 1 C), 143.32 (s, 1 C), 140.66 (s, 1 C), 140.59 (s, 1 C),139.62 (s, 1 C), 133.46 (s, 1 C), 132.06 (s, 1 C), 130.96(s, 1 C), 130.88 (s, 1 C), 129.31 (s, 1 C), 123.88 (s, 1 C),123.85 (s, 1 C), 117.12 (s, 1 C), 115.15 (s, 1 C), 112.79(s, 1 C), 108.71 (s, 1 C), 68.81 (s, 1 C), 64.63 (s, 1 C),64.38 (s, 1 C), 55.68 (s, 1 C), 45.95 (s, 1 C); LCMS[M+H]+= 423.1; HR-MS calcd. for C24H23N2O4[M+H]+:423.2; found:423.172 72 [M+H]+。
2.2.14 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-胺基-2-(4-环丙基甲氧基苯基)丙酰胺(10g)的合成
称量中间体(8) 150 mg 加入反应瓶中,用乙腈40 mL 溶解,25 °C 搅拌下加入溴甲基环丙烷58.6 mg和碳酸铯177 mg,并于40 °C 搅拌反应12 h,TLC显示已达反应终点,加入乙酸乙酯50 mL 后过滤,滤液旋蒸得到中间体(9g),直接用于下步反应。
将得到的中间体(9g)加入反应瓶中,0 °C 搅拌下加入二氯甲烷5.0 mL 溶解,将三氟乙酸1.0 mL 和二氯甲烷4.0 mL 的混合溶液滴加到上述溶液中,并于15 °C 搅拌反应12 h,LC-MS 显示原料已反应完,且发现产物(10g),加入饱和碳酸氢钠10 mL,用二氯甲烷15 mL×3 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(10g),类白色固体50 mg,收率25.4%。1H NMR (400 MHz, DMSO-d6)δ:9.92 (br s, 1H),7.17~7.31 (m, 3H), 6.91~7.01 (m, 1H), 6.87 (br d,J=8.4 Hz, 2H), 6.75 (br d,J= 8.8 Hz, 1H), 4.19 (br d,J=4.4 Hz, 4H), 3.77 (br d,J= 6.8 Hz, 2H), 3.56 (br dd,J=8.4, 5.6 Hz, 1H), 2.98~3.24 (m, 3H), 2.66~2.79 (m, 1H),1.12~1.34 (m, 1H), 0.55 (br d,J= 7.2 Hz, 2H), 0.29 (br d,J= 4.4 Hz, 2H);13C NMR (101 MHz, DMSO-d6)δ:ppm 171.39 (s, 1 C), 158.05 (s, 1 C), 143.31 (s, 1 C),139.59 (s, 1 C), 133.48 (s, 1 C), 131.35 (s, 1 C), 129.22(s, 1 C), 117.14 (s, 1 C), 114.78 (s, 1 C), 112.74 (s, 1 C),108.65 (s, 1 C), 72.40 (s, 1 C), 64.64 (s, 1 C), 64.37 (s, 1 C), 55.47 (s, 1 C), 45.80 (s, 1 C), 10.63 (s, 1 C), 3.56 (s,1 C); LCMS [M+H]+= 369.1; HR-MS calcd. for C21H24N2O4[M+H]+: 369.2; found: 369.164 93 [M+H]+。
2.2.15 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-甲磺酰胺基-2-(4-三异丙基硅氧基苯基)丙酰胺(11)的合成
称量中间体(6) 6.0 g 和三乙胺1.44 g 加入反应瓶中,用二氯甲烷60 mL 溶解,10 °C 搅拌下滴加甲磺酰氯1.30 g,并于25 °C 搅拌反应12 h,TLC 显示已达反应终点,将反应液旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 5/1 to 0/1)纯化得到中间体(11),黄色固体3.90 g,收率75.1%。1H NMR (400 MHz, DMSO-d6)δ: 9.98 (s, 1H), 7.11~7.27(m, 4H), 6.96 (dd,J= 8.8, 2.4 Hz, 1H), 6.79 ~6.87 (m,2H), 6.68~6.77 (m, 1H), 4.15~4.21 (m, 4H), 3.75~3.79(m, 1H), 3.52~3.60 (m, 1H), 3.12~3.17 (m, 1H),3.01~3.02 (m, 1H), 2.83 (s, 3H), 1.18~1.26 (m, 3H),1.04 (d,J= 7.2 Hz, 18H)。
2.2.16 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-甲磺酰胺基-2-(4-羟基苯基)丙酰胺(12)的合成
称量中间体(11) 3.9 g 加入反应瓶中,用四氢呋喃40 mL 溶解,0 °C 搅拌下滴加四丁基氟化铵(1M)7.11 mL,并于0 °C 搅拌反应0.5 h,TLC 显示已达反应终点,反应液用乙酸乙酯200 mL 和纯化水50 mL 萃取,有机层用无水硫酸钠干燥,过滤旋蒸后得到残余物,残余物通过柱层析(石油醚/乙酸乙酯 = 5/1 to 0/1)纯化得到中间体(12),白色固体2.2 g,收率78.9%。1H NMR (400 MHz, DMSO-d6)δ: 9.93(s, 1H), 9.35 (s, 1H), 7.25 (d,J= 2.4 Hz, 1H), 7.12~7.19 (m, 3H), 6.96 (dd,J= 8.8, 2.4 Hz, 1H), 6.74 (br d,J= 8.8 Hz, 1H), 6.71 (br d,J= 8.4 Hz, 2H), 4.14~4.22(m, 4H), 3.72 (dd,J= 9.2, 5.6 Hz, 1H), 3.54 (ddd,J=13.2, 9.2, 6.0 Hz, 1H), 3.06~3.15 (m, 1H), 2.83 (s, 3H)。
2.2.17 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-甲磺酰胺基-2-(4-(4-硝基苯甲酰氧基)苯基)丙酰胺(13a)的合成
称量中间体(12) 100 mg 和4-硝基苯甲酸55.4 mg 加入反应瓶中,用吡啶2 mL 溶解,0 °C 搅拌下滴加三氯氧磷78.2 mg,并于0 °C 搅拌反应3 h,LC-MS 显示原料已反应完,且发现产物(13a),加入饱和碳酸氢钠2 mL,用乙酸乙酯20 mL 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(13a),黄色固体28.0 mg,收率19.6%。1H NMR (400 MHz, DMSO-d6)δ:10.11 (br s, 1H), 8.29~8.49 (m, 4H), 7.49 (br d,J= 7.6 Hz, 2H), 7.21~7.42 (m, 4H), 6.99 (br d,J= 8.0 Hz, 1H),6.77 (br d,J= 8.4 Hz, 1H), 4.20 (br s, 4H), 3.95 (br s,1H), 3.64 (br s, 1H), 3.15~3.27 (m, 1H), 2.88 (br s, 3H);13C NMR (101 MHz, DMSO-d6)δ: ppm 169.78 (s, 1 C),163.65 (s, 1 C), 151.09 (s, 1 C), 150.04 (s, 1 C), 143.38(s, 1 C), 139.88 (s, 1 C), 136.52 (s, 1 C), 134.88~134.95(m, 1 C), 133.17 (s, 1 C), 131.76 (s, 1 C), 129.51 (s, 1 C),124.47 (s, 1 C), 122.28 (s, 1 C), 117.20 (s, 1 C), 112.89(s, 1 C), 108.83 (s, 1 C), 64.65 (s, 1 C), 64.40 (s, 1 C),52.69 (s, 1 C), 45.35~46.15 (m, 1 C), 40.69~40.75 (m, 1 C); LCMS [M-H]+=540.1; HR-MS calcd. for C25H23N3O9S[M-H]+: 540.1; found: 540.109 73 [M-H]+。
2.2.18 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-甲磺酰胺基-2-(4-(4-硝基苯乙酰氧基)苯基)丙酰胺(13b)的合成
称量中间体(12) 100 mg 和4-硝基苯乙酸60.0 mg 加入反应瓶中,用吡啶2 mL 溶解,0 °C 搅拌下滴加三氯氧磷78.2 mg,并于0 °C 搅拌反应2 h,反应液变成棕色,LC-MS 显示原料已反应完,且发现产物(13b),加入饱和碳酸氢钠2 mL,用乙酸乙酯20 mL 萃取,有机层用无水硫酸钠干燥,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(13b),黄色固体28.0 mg,收率19.8%。1H NMR(400 MHz, DMSO-d6)δ:10.06 (s, 1H), 8.24 (m,J= 8.8 Hz, 2H), 7.68 (m,J= 8.4 Hz, 2H), 7.42 (m,J= 8.4 Hz,2H), 7.25 (br d,J= 2.0 Hz, 2H), 7.09~7.20 (m, 2H),6.96 (dd,J= 8.8, 2.0 Hz, 1H), 6.76 (d,J= 8.8 Hz, 1H),4.12~4.24 (m, 6H), 3.89 (br dd,J= 8.8, 6.0 Hz, 1H),3.55~3.67 (m, 1H), 3.16~3.27 (m, 1H), 2.85 (s, 3H);13C NMR (101 MHz, DMSO-d6)δ: ppm 169.78 (s, 1 C),169.74 (s, 1 C), 150.04 (s, 1 C), 147.18 (s, 1 C), 143.36(s, 1 C), 142.31 (s, 1 C), 139.86 (s, 1 C), 136.14 (s, 1 C),133.15 (s, 1 C), 131.51 (s, 1 C), 129.40 (s, 1 C), 123.93(s, 1 C), 122.18 (s, 1 C), 117.19 (s, 1 C), 112.86 (s, 1 C),108.80 (s, 1 C), 64.64 (s, 1 C), 64.39 (s, 1 C), 52.66 (s, 1 C), 45.71 (s, 1 C), 40.69~40.75 (m, 1 C); LCMS [M-H]+= 554.0; HR-MS calcd. for C26H25N3O9S [M+H]+: 556.1;found: 556.138 19 [M+H]+。
2.2.19 N-(2, 3-二氢-1, 4-苯并二氧六环-6-基)-3-甲磺酰胺基-2-(4-环丙甲氧基苯基)丙酰胺(13c)的合成
称量中间体(12) 150 mg 加入反应瓶中,用乙腈40.0 mL 溶解,25 °C 搅拌下加入溴甲基环丙烷61.9 mg 和碳酸铯187 mg,并于40 °C 搅拌反应12 h,LC-MS 显示原料已反应完,且发现产物(13c),反应液中加入乙酸乙酯20 mL,过滤旋蒸得到残余物,残余物通过制备型HPLC 得到目标产物(13c),白色固体31.0 mg,收率18.0%。1H NMR (400 MHz,DMSO-d6)δ:9.96 (s, 1H), 7.15~7.32 (m, 4H),6.82~6.99 (m, 3H), 6.74 (d,J= 8.8 Hz, 1H), 4.18 (q,J=4.8 Hz, 4H), 3.71~3.84 (m, 3H), 3.42~3.65 (m, 1H),3.09~3.19 (m, 1H), 2.84 (s, 3H), 1.13~1.26 (m, 1H),0.48~0.61 (m, 2H), 0.24~0.35 (m, 2H);13C NMR (101 MHz, DMSO-d6)δ: ppm 170.28 (s, 1 C), 158.33 (s, 1 C),143.32 (s, 1 C), 139.70 (s, 1 C), 133.32 (s, 1 C), 130.24(s, 1 C), 129.27 (s, 1 C), 117.19 (s, 1 C), 114.90 (s, 1 C),112.72 (s, 1 C), 108.64 (s, 1 C), 72.44 (s, 1 C), 64.63 (s,1 C), 64.37 (s, 1 C), 52.44 (s, 1 C), 45.77 (s, 1 C), 39.81~39.87 (m, 1 C), 10.61 (s, 1 C), 3.55 (s, 1 C); LCMS[M+H]+= 447.2; HR-MS calcd. for C22H26N2O6S [M+H]+:447.2; found: 447.161 70 [M+H]+。
2.2.20 化合物ROCK2 活性测定
利用Echo 移液系统进行化合物稀释,化合物起始浓度为10 μM,4 倍梯度稀释,11 个浓度点,每个浓度两个重复,按照预先设计的布局,用Echo移液系统将每个浓度的化合物中的100 nL 转移到检测板相应的孔中,在1 000 r·min-1下离心15 s。用40 mM Tris,7.5、20 mM MgCl2、0.1 mg·mL-1BSA 和50 μm DTT 配制ROCK2 激酶缓冲液。用激酶缓冲液准备含0.4 μg·μL-1S6K 底物和20 μM ATP 的混合液,加2.5 nL 的混合液到实验板相应孔中,振荡实验板15 s,在1 000 r·min-1下离心15 s。用激酶缓冲液稀释ROCK2 酶浓度至0.12 μg·mL-1,将该ROCK2 酶溶液2.5 μL 到实验板相应孔(5 μL 反应体系中终浓度为0.3 ng ROCK2, 10 μM ATP 和 0.2 μg·μL-1S6K 底物)。室温23 ℃下,孵育60 min。加5 μL ADP-Glo™ 试剂至实验板相应孔中, 振荡实验板15 s,然后1 000 r·min-1,离心15 s,将实验板置于23 ℃ 孵育40 min。加入10 uL 激酶检测试剂,振荡实验板15 s,然后1 000 r·min-1,离心15 s,将实验板置于23 ℃ 孵育30 min。Envision 读取实验板的发光信号,并加以分析。
3 结论
根据本研究的活性数据,总结目标化合物ROCK2 活性构效关系如下:1)由于10a-d、10g 和13a-c 化合物的ROCK2 抑制剂活性一般,同时参考Netarsudil 和化合物II,说明在药效团的骨架结构中A 环为脂肪环时,会导致活性下降的非常明显,A环为芳香环时会保持较高的活性;2)B 环为脂肪环时同样会导致活性的下降,B 环为芳香环时活性较好,药效团的A 环和B 环,最好为两个相邻的芳香环组成,与靶点蛋白形成疏水作用,会具有很好的ROCK2 抑制剂活性;3)化合物13a-c 的伯胺被甲磺酰基取代后的活性一般,说明化合物的侧链结构中,最好有伯胺基团,其氨基上的氢可以和靶点蛋白形成较强的氢键,使活性增高;4)侧链中含有芳香环的10e 和10f 具有一定的活性,而脂肪环10b、10c 和10g 的活性结果一般,说明侧链的结构中最好有一个芳香环,且该芳香环在3 位和6 位有取代基的话,会增强活性。本研究结果同时显示化合物10e 和10f 表现出了一定的体外ROCK2 酶抑制剂活性。接下来的研究会进一步按照该构效关系,以化合物10e 和10f 为基础设计新的药效团骨架,试图找到具备有进一步开发价值的可用于治疗青光眼的新型ROCK2 抑制剂。
