环境DNA宏条形码技术在蓝藻群落监测中的应用
2021-03-02李小闯霍守亮张含笑金小伟李文攀张军毅张靖天
李小闯, 霍守亮*, 张含笑, 金小伟, 李文攀, 张军毅, 张靖天
1.中国环境科学研究院, 环境基准与风险评估国家重点实验室, 北京 100012 2.中国环境监测总站, 北京 100012 3.无锡市环境监测中心站, 江苏 无锡 214023
水体富营养化和气候变暖背景下,蓝藻水华暴发频率、范围、周期在不同程度上有所增加[1-2]. 部分水华蓝藻产生藻毒素和异味物质,严重威胁饮用水源地水质安全,限制水体的生态服务功能,对社会经济发展产生重要影响[3-4],迫切需要大规模和长周期的蓝藻群落监测. 基于形态特征的显微镜镜检方法耗时、耗力,无法满足大量样品观测需求. 另外,镜检需要熟悉蓝藻分类系统并具备样品观察经验的专业人员,分析结果因个人主观经验的干扰而差异较大. 有研究让两位分类学家同时观察17株蓝藻培养物,发现仅3株在种水平鉴定结果一致,6株在属水平结果一致,7株鉴定为不同的属[5]. 长期项目或跨区域合作项目往往需要多个藻类分析员,保持监测结果一致性和可比性存在困难. 另外,镜检容易忽视超微藻类群(粒径<2 μm),当环境中蓝藻未形成典型形态特征时,如厚壁孢子、异形胞、末端特殊结构等,往往无法鉴定到种水平或出现鉴定错误[6-7].
蓝藻的原核特征被发现之前,一直被认为是真核藻类,物种命名以形态特征为基础,遵循植物学命名系统. 直至20世纪60年代,由于电镜和分子信息等技术发展,发现蓝藻属于原核生物[8]. 细菌学者认为,蓝藻应采用细菌学命名系统,物种命名基于模式菌株(纯培养物)的观察而确定[9]. 蓝藻的原核特征被发现后,蓝藻的命名采用植物学分类系统或细菌学分类系统. 同一蓝藻物种在两个分类学系统下描述和命名,一物种两学名给蓝藻分类系统造成了困扰. 20世纪80年代以来,Anagnostidis和Komrek提出多相分类学方法(polyphasic approach),融合形态特征、超微结构、繁殖方式、生理生态特征和分子数据等进行综合分析,逐步完善了蓝藻分类系统[10-11],形成经典的蓝藻四目系统(色球藻目、颤藻目、念珠藻目、真枝藻目),该分类系统流传广、接受程度高. 2000年以来,分子数据以指数倍增长,通过分子系统发育分析对基于形态特征描述的蓝藻分类系统进行校正和修订,大量新的蓝藻种属被描述和确立. 更高层次的蓝藻分类单元(如色球藻目和颤藻目)并非单系类群,Komárek等[12]基于形态特征、类囊体结构以及分子数据对四目系统进行修订,提出了八目系统,即粘杆菌目、聚球藻目、螺旋藻目、色球藻目、宽球藻目、颤藻目、拟甲色球藻目、念珠藻目.
形态特征是基因表达的外化,是基因和环境共同作用的结果. 基于形态特征描述的蓝藻分类系统不能反映各类群间进化关系,形态特征相似的物种可能进化上起源不同,而形态特征差异大的物种可能起源相同[13]. 分子数据的应用可以描摹出蓝藻类群间进化关系,分子系统发育分析是现代蓝藻分类学修订中最重要的方法之一. 基于分子特征修订的蓝藻分类系统可以克服基于形态特征的弊端,更能准确地揭示蓝藻物种多样性组成.
2000年后,以高测序通量为显著优势的二代测序技术发展快速,高效的生物信息学处理方法和软件相继研发,进一步推进了eDNA宏条形码技术用于蓝藻群落监测的探索研究,以获得蓝藻群落组成、物种丰度及反映物种间进化关系的注释结果. 基于分子特征修订的蓝藻分类系统还未完善,处于持续修订中,但eDNA宏条形码为快速和大规模蓝藻群落监测提供了可能,有必要推进eDNA宏条形码在蓝藻群落监测中的应用. eDNA宏条形码在蓝藻群落监测中应用处于初步探索阶段,该研究从引物选择、序列聚类、注释方法和绝对定量4个层面概括宏条形码数据处理方法,并对eDNA宏条形码技术在蓝藻群落监测中的应用提出了参考建议,以推进eDNA宏条形码在蓝藻群落监测中的应用.
1 eDNA宏条形码技术内涵及在蓝藻群落监测中的应用
eDNA指从环境样本中提取的DNA,eDNA宏条形码指利用特定分子标记对eDNA进行扩增,扩增产物进行高通量测序获得DNA序列,通过序列检索比对鉴定多个物种,广义上宏条形码不仅限于高通量测序. 简单来说,eDNA宏条形码是指利用特定分子标记检测eDNA来同时鉴定多个物种. eDNA宏条形码一次产生数万条以上目标基因扩增子序列,由于测序通量大,蓝藻多样性远高于镜检方法,不仅可以揭示蓝藻群落组成,还有助于发现新物种[14]. 宏条形码技术能实现大批量样品的同时处理,试验操作流程可标准化和自动化,分析结果不因操作人员或不同实验室而异,有利于高效、准确地获得大规模蓝藻监测数据,为蓝藻生物地理学分布特征研究提供可能[15].
eDNA宏条形码已开始应用在蓝藻群落监测,涉及湖、库、河流、极地融雪水体、以及湖库沉积物[16-18]. 一些研究关注特殊生境蓝藻群落,如沙漠结皮、湖底或岸边蓝藻席、超微蓝藻等[13,19-20]. eDNA宏条形码检测蓝藻多样性显著高于镜检法,且两种方法检测的物种数目呈显著相关,eDNA宏条形码在蓝藻群落监测中有较高的优势[18]. 用扩增子深度测序的方法对我国3个富营养状态湖库蓝藻群落开展研究,发现微囊藻(Microcystis)是优势类群,但一定环境条件下,聚球藻(Synechococcus)在水面形成优势类群[16]. 聚球藻是超微藻类,在淡水和海洋常见,物种多样性高,适应多种环境条件[21],是潜在的蓝藻水华形成物种. 常规监测中超微藻类易被忽略,考虑到聚球藻具有发生蓝藻水华的潜在态势,超微藻类应纳入为一项常规监测指标.
eDNA宏条形码用于南极洲雪融水体栖生蓝藻席研究,发现蓝藻多样性较高,绝大多数蓝藻席(cyanobacterial mats)存在微囊藻毒素和拟柱孢藻毒素[19]. 另外,eDNA宏条形码可以实现长时间序列蓝藻群落变化研究,揭示历史蓝藻群落组成和水生态健康状况. Tse等[17]采用eDNA宏条形码和古湖沼学技术探究加拿大一水库蓝藻群落百年尺度变化,发现近几年长孢藻(Dolichospermum)相对丰度增加,与微囊藻毒素基因(mcyA)基因拷贝数变化趋势相同.
2 eDNA宏条形码技术在蓝藻群落监测中的方法进展和关键技术
2.1 引物选择
eDNA宏条形码技术需要构建分子标签扩增子数据库,扩增产物一般要求低于500个碱基对. 扩增引物遵循以下两个原则:①选定基因序列易于PCR扩增,且方便设计通用引物;②选定条形码区域能凸显种间差异,但种内差异小[22].
核糖体小亚基16S核糖核酸基因(16S small ribosomal subunit RNA gene, 16S rRNA gene)是蓝藻多样性研究应用最广的基因,既有高度保守性区域,又有多个高度变异性区域,可同时用于反映物种间的亲缘关系和物种间的差异[23]. 近年来,GenBank数据库蓝藻16S rRNA 基因序列呈指数趋势增加(见图1),提升了蓝藻分子鉴定的精确度和分子系统发育构建的稳健性,以及评估引物的特异性. 二代测序受测序读长限制,只能选择1~3个可变区作为扩增片段,不同可变区突变点位分布不均,选择的扩增片段不同往往导致物种的分子鉴定结果出现偏差. 三代测序集合了Sanger测序的读长优势和二代测序的高通量优势,可用较低的成本获得16S rRNA 基因全长序列,避免了因选择不同可变区域为扩增片段而影响蓝藻物种分子鉴定结果的准确性. 16S rRNA 基因全长测序用于对23个培养菌株形成的模拟群落研究,发现16S rRNA 基因全长序列鉴定准确度高达99%,且重复性高;更全面的序列信息提高了物种鉴定的准确性,有利于种水平物种鉴定,对系统发育、群落鉴定和代谢通路预测研究更有优势[24].
16S rRNA基因在结构和功能上较保守,物种间16S rRNA基因序列差异小,不适合在物种水平上进行区分[25]. 16S-23S rRNA基因间隔区域(internal transcribed spacer, ITS)序列长、变异大,属内物种间差异显著[26]. 基于ITS基因系统发育分析,拟圆孢藻属(Sphaerospermopsis)3个物种各自形成单系类群,能彼此区分[27]. 相对核基因,功能基因进化快、含有变异信息多,能反映物种适应环境变化的生理生态特征,已被广泛用于寻找物种间差异,比较常用的基因包括依赖于脱氧核糖核酸的核糖核酸聚合酶基因(DNA-dependent RNA polymerase gene,rpoC1)、藻蓝蛋白及其转录间隔区(phycocyanin operon with intergenic spacer region,cpcBA-IGS)等[28]. 微囊藻是全球最普遍的水华蓝藻物种,据AlgaeBase数据库统计已有113个形态种,但形态物种间区分鲜少得到分子证据的支持. 基于cpcBA-IGS基因系统进化分析,惠氏微囊藻(M.wesenbergii)形成一独立进化枝,可以和其他微囊藻物种区分开[29]. 功能基因有利于属内物种间区分,应继续探究其他功能基因在形态物种间区分的分子证据. 笔者基于GenBank数据库统计,认为目前ITS间隔区及功能基因序列数目显著低于16S rRNA基因序列数目,且不同功能基因序列数目各异,不利于环境样品浮游植物物种精准鉴定(见图1).
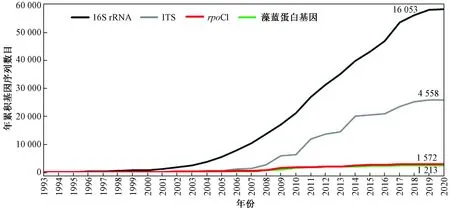
注: 折线上数字代表去除未培养藻株序列数目. 1993年包括1993年以前数据,数据收集截止至2020年4月2日.图1 GenBank数据库蓝藻16S rRNA、ITS、rpoC1、藻蓝蛋白基因序列(>200 碱基对)数目随时间变化量Fig.1 Number of gene sequences larger than 200 base pairs within GenBank for the cyanobacterial 16S rDNA, ITS, rpoC1 and phycocyanin loci over time
蓝藻水华通常由部分关键蓝藻物种引发,探究水华蓝藻种群结构、时空动态变化、基因多样性,解析蓝藻水华演替机制,可为蓝藻水华防控和水质管理提供理论依据. 微囊藻、长孢藻、拟柱孢藻(Cylindrospermopsis)是世界范围内常见的水华蓝藻类群,目前针对3个类群的引物已研发出来并进行基因型和水华种源的挖掘[30]. 通过构建克隆文库发现太湖长孢藻存在两个主基因型,主基因型随着季节变化进行演替,表明不同水华阶段由不同长孢藻生态型组成[31]. 基于高通量测序的eDNA宏条形码检测到微囊藻基因多样性远高于克隆文库,发现的微囊藻多样性和富营养化水平呈负相关[32]. eDNA宏条形码用于水华蓝藻基因型研究鲜见报道.
2.2 序列聚类
eDNA宏条形码数据处理的第一步是将扩增子序列聚类为操作分类单元(operational taxonomic unit, OTU). 通过设定物种间分类阈值,将相似度超过该阈值的序列聚类为相同OTU. 然而分类阈值易使序列信息差异太小的物种被掩盖,导致生物多样性被低估. 近年来,序列差异低至单核苷酸变异的处理方法被提出,克服了物种丢失的弊端,可以检出序列高度相似但属于不同种属的物种和生态型[33].
通过距离度量方法计算不同序列之间的相似性,序列相似性高于分类阈值的聚为同一OTU. 采用OTUs聚类对eDNA宏条形码数据进行处理,不仅缩减工作量,提高分析效率,而且剔除了扩增过程中的嵌合体以及测序过程中高通量测序仪器引入的错误序列[34],提高了分析的准确性. Qiime分析流程提供的OTU聚类方法有3种,包括denovo、closed-reference、open-reference聚类.
无参OTUs聚类(denovoOTUs clustering). eDNA宏条形码序列通过距离度量计算序列之间的相似性,每一序列都需与其余序列比对,比较序列间距离相似度和设定的分类阈值,将上万条序列聚类成OTUs[35]. 数据通量越高,错误测序序列和单碱基变异的序列(unique sequence)增加,分层的无参聚类方法计算代价和时间以几何倍数增长;序列比对不能同时进行,比对数据量大、耗时,适用于小规模数据集分析[36]. 无参聚类根据分类阈值聚类OTUs,可以消除一些错误序列. 对原核生物16S rRNA 基因的eDNA宏条形码研究中,OTUs聚类的分类阈值通常设置为97%[37],后续处理中OTUs被当成物种,用来评价物种多样性. 然而,该分类阈值是人为设定的,序列相似度高于该阈值的不同物种可能被掩盖,不能准确体现物种间的进化关系,OTUs的生物学意义有待校验. 不同的数据集OTUs难以合并分析,不利于不同研究的比较分析.
有参OTUs聚类(closed-reference OTUs clustering). 与无参聚类不同,有参聚类需要参考数据库,通过设定分类阈值,将输入序列和参考序列距离相似度在阈值范围内的扩增序列归为相同OTU[38]. 参考数据库挑选高质量的基因全长序列,参考序列间相似度低于97%,每一参考序列代表不同物种[36]. 有参聚类会出现输入序列和多个参考序列相似度在分类阈值范围内,导致序列注释不明确[39]. 有参聚类会设定聚类中心,序列比对可同时进行,数据处理高效,适用于大数据集分析[40]. 有参聚类产生的OTUs具有生物学意义,不同数据集采用相同的参考数据库,可以进行不同研究的比较分析. 有参聚类不能鉴别新物种,如果输入序列和参考数据库所有序列相似度均低于分类阈值,该序列被舍弃,丢弃了参考数据库以外的新物种. 随着参考数据库不断完善,这一缺点会逐渐弱化.

注: 无参OTUs聚类方法限于自身数据集内,有参OTUs聚类方法不能得到数据集外的生物学变异,扩增子序列变异方法能得到所有的数据和生物学变异[43]. 图2 基于研究数据集的3种序列聚类方法有效性程度Fig.2 The extent of the validity of de novo OTUs, closed-reference OTUs and ASVs methods determined from the study dataset
开放式有参OTUs聚类(open-reference OTUs clustering). 开放式有参聚类融合了无参聚类和有参聚类的优势,输入序列先通过有参聚类方法聚类成OTUs,未匹配序列再通过无参聚类生成OTUs,产生的OTUs具有生物学意义,也保留了数据库以外的新物种[41]. 因无参聚类方法存在,不能避免耗时长的弊端,不利于大数据集处理. 二次抽样开放式有参聚类(subsampled open-reference OTU clustering)是开放式有参聚类方法的优化版本,通过随机抽取与数据库参考序列未匹配的扩增序列,增加为参考数据库序列,序列聚类可并行运行,从而有效地缩短聚类时间,适用于大数据集分析[42].
扩增子序列变异(amplicon sequence variants, ASVs)在引入错误序列之前得到有生物学意义的序列,基于熵值在单核苷酸的精度上区分序列之间的差异,可以用于区分生态型[43]. ASVs的核心思想是具有生物学意义的序列可以和错误序列区分,且数目远大于错误序列数. 错误序列在有效控制下,ASVs的准确度会增加. ASVs代表了具有生物学意义的序列,因此不受数据集的限制,不同的数据集之间可以进行比较分析. ASVs数据分析可重复,能更准确、更全面地评估eDNA宏条形码数据结果. 若物种基因组的基因片段有多个拷贝,会得到多个ASVs,导致该物种区分不明确. 目前,ASVs分析有以下几种方法,包括DADA2[44]、Unoise2[45]、Deblur[46]、Oligotyping[33]、Minimum Entropy Decomposition[47]、Sub-OTU resolution method[48]. 这些方法不需要参考数据库,经过质量控制的序列降噪去除扩增与测序过程中的错误序列,得到具有生物学意义的序列(见图2).
基于Sub-OTU resolution method分析微生物群落,发现基于97%分类阈值聚类的OTU内多达20个亚群落,这20个亚群落生态特征明显不同,序列低至一个核苷酸差异(99.2%相似度)[48]. Oligotyping分析沼泽存在22个远洋杆菌属(Pelagibacter)寡核苷型,其中2个优势寡核苷型序列差异低至一个核苷酸(99.57%相似度),一个寡核苷型在冷季占优势,另一个在暖季占优势,表现不同的季节演替[33]. 低至单核苷酸差异的序列可以表现不同的生态特征,若以分类阈值聚类OTUs可能忽略这些生态型.
2.3 注释方法
序列比对的物种注释方法是通过与数据库参考序列距离相似度比较给予物种名称,注释结果的准确度与参考数据库的选择息息相关. 现有的16S rRNA 基因数据库,如SILVA[49]、RDP[50]、Greengenes[51],由高质量16S rRNA 基因全长序列组成,数据库大多数序列由环境样品扩增而来,序列注释的物种名称多是基于预测获得,许多序列未注释物种名称[52]. 由于参考数据库的完善性,eDNA宏条形码序列比对注释结果存在一定比例未注释的序列[19-20]. 另外,大量蓝藻纯培养物基于形态特征的鉴定结果缺陷,扩增的基因序列提交至数据库[9],若序列未核查和修订,会导致eDNA宏条形码序列错误的注释结果.
基于系统进化位置(phylogenetic placement method)注释的方法可以对参考序列进行修正,提升输入序列注释的准确性,并且反映了输入序列之间的进化关系[53]. 分子数据是现在蓝藻分类学修订最重要的指标之一,蓝藻属的修订和新属的提出需满足进化分枝是单系类群[9-10]. 由于分枝未延伸到属以下的分类单元,没有界定种水平需满足单系类群. 系统进化种(phylogenetic species concept)是系统进化关系能可靠推断出的最小分类单元. 从谱系学的角度来看,系统进化种规定物种产生于进化关系间的转变,可以在系统进化关系上将系谱关系连接起来[54]. 分类学和系统发育分析的结果常常不一致,系统发育分析的结果和序列质量、序列比对质量以及方法和参数息息相关.
基于最大似然法的系统发育分析,是推断未知序列分类学最可靠的方法,由于计算复杂性和缺乏系统进化信号,不适用于大数据分析[55]. 系统进化位置的注释方法,首先基于参考序列构建系统发育树,输入序列通过序列比对添加到参考系统发育树上,该方法可以对数以万计的输入序列快速又准确的注释,相关软件有EPA (evolutionary placement algorithm)[56]和pplacer[55]. EPA和pplacer分析速度和准确度相当,pplacer的参考系统进化树和进化枝是固定的;而EPA优化了参考系统进化树的进化枝长度以及输入序列在进化枝的位置,每一个输入序列的注释推导是基于略有差异的参考系统进化树.
2.4 绝对定量
eDNA宏条形码分析往往仅考虑物种的相对丰度,然而相对丰度不能反映样本中真实的绝对丰度. 相对丰度相同的样本绝对丰度可能差别很大,相对丰度差别很大的样本绝对丰度可能相同,因此相对丰度可能导致错误的预估结果,掩盖了生理学和生态学潜在的有价值的信息[57-58]. 相对丰度不能体现物种在不同样本中的绝对含量的变化,不利于不同样本间的比较分析. 定量PCR常用于获得样本微生物总丰度,但分析结果因DNA提取和扩增效率的不同而出现偏差[59-60]. 以细菌基因组或人工合成的嵌合体DNA作为内参的内标法[58,61],以及利用细胞生物量和拷贝数间关系的细胞体积校正系数法[62]等绝对定量方法,为跨样本间物种丰度的比较分析提供了可能.
内标法是在样本微生物基因组提取之前,将内标DNA添加到样本中,与样本微生物共提取基因组并进行扩增,根据内标DNA丰度推算样本微生物总丰度. 内标DNA可以来源于细菌[61,63],或者合成的嵌合DNA[58]. 内标DNA的加入量应根据样本DNA总量来决定,在0.1%~1.0%之间均可,加入量既要保证有足够的内标序列来分析,也要避免加入太多造成对原始微生物群落的检测带来影响[61]. 细菌应选取不存在于样本群落中且与样本中任何普遍存在的物种相似度都不高,避免对样本中微生物绝对定量分析的影响. 合成的嵌合DNA可以弥补这一缺陷,嵌合DNA设计包含以下3个关键要素:①目标研究类群基因的引物结合位点;②与扩增产物相同长度和GC含量的优化合成填充序列;③合成嵌合DNA的来源易于获得. 样本中可同时加入针对多个类群(细菌、真菌、真核生物)的嵌合DNA,能一次实现多个类群的绝对定量,可以在生物域水平揭示菌群结构的变化〔见图3(a)〕.
内标法只能对样本微生物总基因拷贝数进行绝对定量,不能直接获得微生物绝对丰度. 每种微生物细胞含有一到多个16S rRNA基因拷贝数,扩增后测序会放大这种基数效应,造成物种的序列数产生偏差. 虽然rrnDB在线数据库可以基于物种全基因组对基因拷贝数进行评估和矫正来获取eDNA宏条形码数据每个物种绝对丰度[64],但现仅有400个蓝藻藻株基因组,且大多数藻株来自于海洋和法国巴斯德研究所保存的淡水蓝藻株,只能覆盖极少数蓝藻物种[65]. 因此,基于rrnDB数据库只能对部分蓝藻物种基因拷贝数进行矫正,且校正结果因不同研究区域而有所偏差.
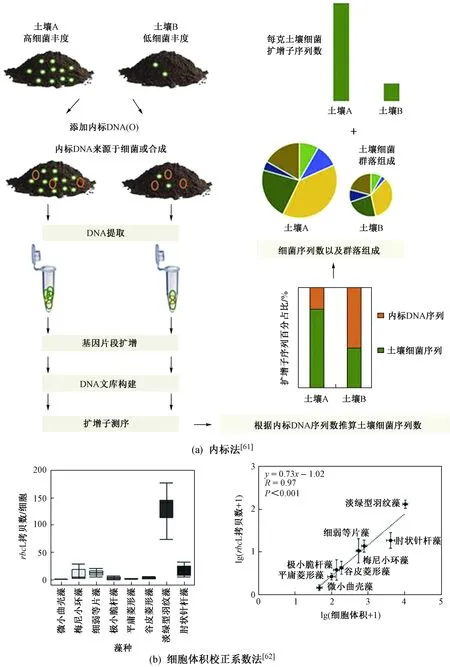
图3 eDNA宏条形码绝对定量方法[61-62]Fig.3 Methods of absolute quantification of eDNA metabarcoding[61-62]
细胞体积校正系数法(correction factor, CF)是通过物种细胞体积和细胞含有的基因拷贝数目之间建立联系得到校正系数,实现eDNA宏条形码物种丰度的绝对定量. 该方法已对环境鱼类、两栖动物[66]、寡毛纲[67]、细菌[68]、古菌[68]以及硅藻[62]进行评估和定量,而关于蓝藻类群的研究还鲜见报道.
Vasselon等[62]发现硅藻8个物种细胞体积、长度、宽度和rbcL拷贝数显著相关. 基于8个物种混合类群评估,eDNA宏条形码数据经过CF校正和显微镜镜检得到物种绝对丰度结果一致,而未经CF校正的丰度和镜检数据存在很大差异〔见图3(b)〕. 基于环境硅藻类群评估,经过和未经CF校正的eDNA宏条形码数据,优势类群发生明显变化,表明eDNA宏条形码数据未经过CF校正直接影响硅藻群落结构变化,如大体积物种丰度被高估,而小体积物种丰度被低估,这一变化导致了生态学意义的差异[62]. 经过CF校正的环境样本硅藻和寡毛类绝对丰度,由其推导的水质评价结果和镜检结果是可比的,可以替代形态监测方法进行水体生态评价[62,67].
CF假定细胞基因拷贝数和细胞体积是稳定的,但它们会因生理状态的不同而发生变化,如不同环境和生活周期等[69]. 虽然细胞体积因环境条件而改变,但Mann[70]发现环境水体硅藻细胞大小分布是呈正态分布的,大、小个体数目均衡克服了这一缺陷. 未来研究需考虑细胞基因拷贝数和细胞体积的变化对CF的影响;另外,还需评估更多物种细胞体积和基因拷贝数之间联系,以获得更准确的CF值.
3 结论与展望
eDNA宏条形码因通量高、易发现新物种,相比形态方法,可以更准确地揭示微生物群落结构和组成. eDNA宏条形码已经应用于探究各种生境蓝藻群落组成,检测物种多样性远高于显微镜检法,群落组成可以反映不同季节和不同富营养化程度水体之间差异,研究发现聚球藻等超微藻类可能是潜在的水华发生类群,eDNA宏条形码在蓝藻群落监测和水生态健康评价中表现出极大的优势性. 但以往研究在eDNA宏条形码数据处理过程中未考虑生态型且注释结果未反映物种间的进化关系,导致潜在的生理学和生态学信息被掩盖,不利于对水体生态健康进行科学评价和管控. 因此,笔者从以下几个方面对eDNA宏条形码技术在蓝藻群落监测的数据处理方法优化提出了建设性意见,以准确地揭示蓝藻群落结构和组成.
a) 引物选择. 针对蓝藻,引物选择应能覆盖广谱的蓝藻物种;针对水华蓝藻类群以及产毒、产异味类群,选择合适的基因并对设计引物进行测试. 目前16S rRNA 基因是eDNA宏条形码最常用的选择,因该基因数据库涵盖蓝藻物种范围广. 但16S rRNA基因在属内物种间区分效果不好; 再者,16S rRNA基因数据库含有部分物种名称注释错误的序列,导致eDNA宏条形码序列注释错误. 应对数据库16S rRNA基因序列进行修正,追溯到原始发表文章进行基于形态特征鉴定的核验,为eDNA宏条形码序列准确的注释提供高质量源头序列. ITS基因以及功能基因变异大、含有更多序列变异信息,在蓝藻属内物种间具有较好的区分,但基因数据库体量小,未来研究应扩充数据库ITS基因以及功能基因序列,为eDNA宏条形码在蓝藻物种水平注释提供可能.
b) 单核苷酸变异序列具有生物学意义. OTUs聚类通过设定物种间分类阈值且序列相似度高于该阈值的序列聚类为相同OTU,然而序列信息差异过低的物种和生态型可能被忽视,导致生物多样性被低估. ASVs可以在单核苷酸的精度上区分序列,用于区分生态型,ASVs代表了具有生物学意义的序列,实现了跨数据集间的比较分析.
c) 基于系统进化位置的注释方法反映物种间的进化关系. 系统进化位置方法不仅可以对参考数据库序列进行修正,提升输入序列注释的准确性,另外也反映了序列间进化关系. 系统进化位置方法能同时对数以万计的输入序列快速又准确的注释,注释准确度和分辨率随着数据库物种多样性增加和蓝藻分类学系统的持续修订及完善而提升.
d) 绝对定量. 相对丰度不能反映样本中真实的绝对丰度,相对丰度和绝对丰度可能存在巨大反差,因此基于样本相对丰度分析可能导致错误的预估结果,掩盖了生理学和生态学潜在的有价值的信息. 内标DNA法和细胞体积校正系数法为eDNA宏条形码用于蓝藻丰度的绝对定量提供了可能,为物种丰度的跨样本间分析提供了可能.
