内质网应激反应IRE1α信号通路与细胞衰老在癌症中的作用及关系的研究进展
2021-02-22郭茜晨张婷婷王润新赵法明
郭茜晨,张婷婷,王润新,赵法明,盛 夏
(华中科技大学同济医学院公共卫生学院环境医学研究所,湖北 武汉 430030)
内质网是细胞内负责蛋白质加工转运的主要场所。当蛋白质由于各种原因不能正常折叠成高级结构而在内质网腔中积累时,会导致内质网应激(endoplasmic reticulum stress,ERS),激活未折叠蛋白反应(unfolded protein response,UPR)。UPR通过暂停蛋白质转录和翻译、降解错误折叠的蛋白质、激活分子伴侣表达从而参与蛋白折叠等多种方式缓解内质网应激。在癌症发展过程中,肿瘤细胞常常由于所处微环境引起内质网应激,UPR在肿瘤细胞的生存和适应应激条件中起着关键作用。
细胞衰老是一种细胞周期停滞的永久状态。DNA损伤、癌基因的激活、氧化应激、化疗、线粒体功能障碍等原因都会导致细胞衰老现象的发生。衰老的细胞表现出结构上的畸变,表现为细胞形态变大、变平、质膜成分改变、溶酶体和线粒体的积累以及细胞核的改变。其表型通常表现为激活慢性DNA损伤反应(DNA damage response,DDR)、参与多种细胞周期蛋白依赖性激酶抑制剂(cyclin-dependent kinase inhibitors,CDKi)的形成、促进促炎症因子和组织重塑因子的分泌、诱导抗凋亡基因、改变代谢率和内质网应激。
随着细胞的逐渐衰老,氧化应激增加,受损的蛋白质在内质网腔中逐渐积累,引发UPR。同时,细胞衰老也会促进增生性病变,癌症便是其中重要表现之一。肌醇依赖性激酶1α(inositol requiring enzyme1α,IRE1α)信号通路作为研究最广泛的UPR信号网络分支,其在癌症与细胞衰老中的作用已被广泛证实。鉴于此,本综述归纳了IRE1α信号通路在常见癌症中的作用,细胞衰老与癌症的关系,以及IRE1α信号通路与细胞衰老在癌症发展中的相互作用,旨在为肿瘤治疗提供新的方向。
1 IRE1α信号通路
UPR信号网络由3个跨内质网膜的感应蛋白所介导,分别为IRE1α,蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)和激活转录因子6(activating transcription factor 6α,ATF6α)。正常状态下,这3种蛋白的内质网结构域与分子伴侣(glucose regulated protein78,GRP78)结合并处于静息状态。当发生内质网应激时,GRP78主动结合腔内聚集的未折叠蛋白并与UPR感应蛋白分离,IRE1α、PERK和ATF6α这3条经典的信号传导通路随之被激活。
IRE1α通路是UPR信号网络中进化上最保守的分支。IRE1α是一种相对分子质量为110 ku的I型跨膜蛋白,其C端位于胞浆,具有蛋白激酶和核糖核酸内切酶双重活性。内质网应激时,IRE1α与GRP78解离后发生二聚化和自磷酸化,其RNase活性被激活,介导XBP1 mRNA的剪切,进而翻译生成转录因子XBP1s。XBP1s的靶基因包括p58IPK
、ERdj4
、HEDJ
等编码伴侣蛋白的基因,EDEM
等与内质网相关降解(endoplasmic reticulum associated degradation,ERAD)相关的基因,RAMP4
等涉及糖基化的基因,来调节蛋白质折叠与分泌,促进蛋白质向内质网转运和脂质合成,促进ERAD来缓解内质网应激。激活的IRE1α还可以降解特定的内质网结合的mRNA,这一过程被称为调节的IRE1依赖性衰变(regulated IRE1α-dependent decay,RIDD)。RIDD有助于减少进入内质网的新生蛋白质的折叠负荷,从而进一步缓解内质网应激。当内质网应激无法缓解时,IRE1α还可以激活JNK信号通路,介导细胞凋亡。IRE1α通路的激活和下游信号通路详见图1。综上所述,IRE1α-XBP1s通路在内质网应激与细胞命运决定中发挥重要作用。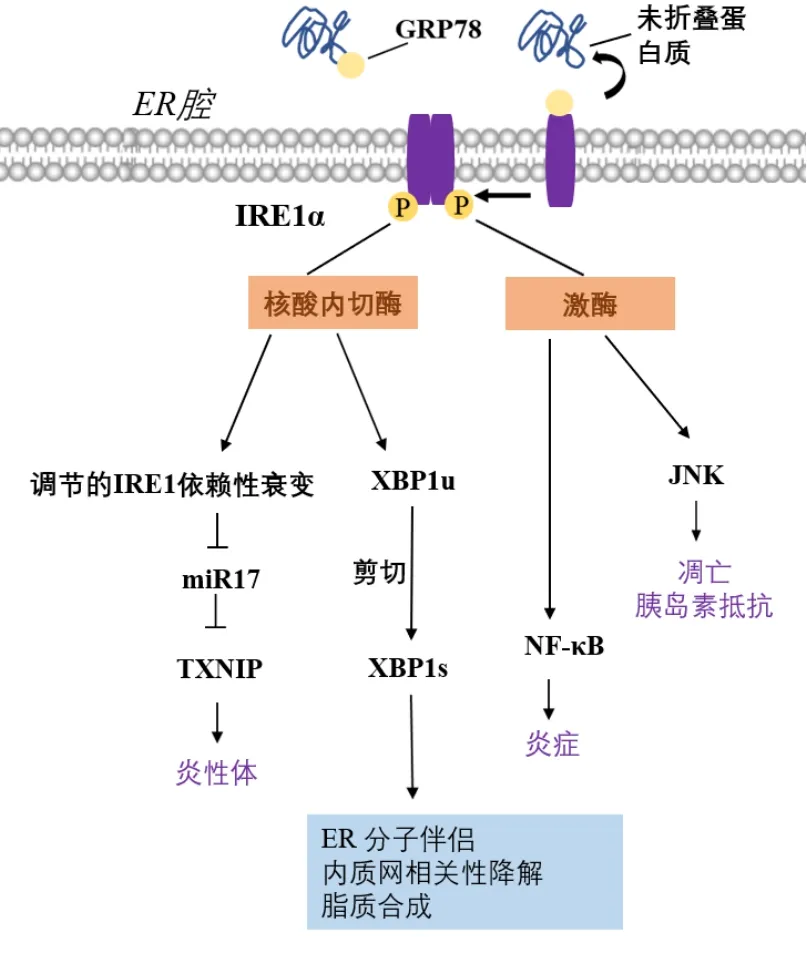
图1 UPR的IRE1α信号通路
2 IRE1α信号通路在癌症中的作用
癌症的形成涉及多种复杂的细胞应激反应,内质网应激时常发生。UPR作为缓解内质网应激的有效手段,对癌症的发展具有推动作用。IRE1α信号通路作为UPR研究中最为广泛的通路,在多种癌症进展中发挥重要作用。XBP1剪切产物XBP1s可以通过p53诱导,激活NF-κB、AP(activator protein)和MYC等介导的多种致癌途径,还可以通过调节PI3K/mTOR信号通路活性,促进癌症的发生发展。IRE1α信号通路在一些常见癌症中的作用总结如下。
2.1 肝癌
研究表明,XBP1s在肝癌细胞中的表达量显著高于正常细胞,且肝癌组织中XBP1s蛋白的表达水平也高于正常肝组织。此外,肝脏特异性IRE1
α基因敲除可降低肝癌模型小鼠的发病率,并减缓肝癌的发展。与周围的非肿瘤组织相比,GRP78在肝细胞癌患者的肿瘤样本中被发现存在组成性过度表达,且其表达与XBP1剪接相关。由XBP1s在肝癌组织中的表现可见IRE1α信号通路与肝癌发展密切相关。2.2 乳腺癌
在乳腺癌中XBP1s被证明可以与HIF1α相互作用,协同促进肿瘤血管生成。有报道表明,XBP1s在三阴性乳腺癌中被激活,并在该类乳腺癌亚型的致瘤性和进展中起关键作用。在乳腺癌细胞系模型中,XBP1s转录调控网络的全基因组图谱显示,XBP1s可以通过与HIF1α组装转录复合物来驱动三阴性乳腺癌的致瘤性。XBP1s还能通过其他的途径影响乳腺癌细胞的生长。MYC蛋白可以通过与IRE1α启动子和增强子区域结合促进IRE1α-XBP1s通路的表达,并且MYC
基因也可直接与XBP1s相互作用,进而调控XBP1s转录活动,促进肿瘤细胞的生长。若使用小分子化合物MKC8866可特异性抑制IRE1α核酸内切酶活性,降低XBP1s的合成,从而选择性抑制患者肿瘤的异种移植和基因工程小鼠模型中MYC过表达肿瘤的生长。以上所述的XBP1s与HIF1α及MYC之间的作用提示IRE1α-XBP1s通路在乳腺癌中发挥重要作用。2.3 前列腺癌
研究表明在前列腺癌细胞中,雄激素受体与UPR相关的基因调控位点结合,可在转录水平激活IRE1α分支,而IRE1α-XBP1s通路通过激活c-Myc信号来促进前列腺癌的发展。与前述一致,IRE1α核酸内切酶抑制剂MKC8866能够有效抑制多种人源前列腺癌细胞在动物模型中的生长,并能与二代雄激素信号抑制药物恩杂鲁胺Enzalutamide产生协同作用。XBP1s特异性基因表达产物也与前列腺癌的较短的无病生存期之间存在显著关联。此外,最新的研究也表明IRE1α能够与MYC形成合成致死效应,而在MYC高表达的肿瘤中靶向IRE1α,可能与抑制MYC具有协同作用。靶向UPR已被提出为治疗MYC过度激活的肿瘤的一种新治疗策略。
2.4 卵巢癌
IRE1α-XBP1信号通路可以通过调节线粒体活性控制卵巢癌T细胞功能。研究表明从卵巢癌患者标本中分离的T细胞中,XBP1的上调与T细胞向肿瘤的浸润减少和干扰素IFNG mRNA表达减少有关。肿瘤相关树突状细胞(tumor-associated dendritic cells,tDCs)中XBP1的激活可通过减弱抗肿瘤免疫来驱动卵巢癌的进展。这是因为脂质过氧化副产物会引发XBP1活化,并在肿瘤相关树突状细胞中诱导甘油三酯生物合成程序,导致异常的脂质积聚,随后抑制了肿瘤相关树突状细胞支持抗肿瘤T细胞的能力。此外,与正常卵巢上皮细胞相比较,浆液性卵巢癌细胞中XBP1的表达上调。在XBP1
选择性敲低的细胞群组中,丝裂原活化蛋白激酶MAPK p38的磷酸化水平呈现下降趋势,卵巢癌细胞增殖活力下降。这些结果表明,XBP1可能在浆液性卵巢癌细胞中起到抗氧化应激的保护作用,其潜在的分子机制或与磷酸化p38的下调相关。此外,XBP1也可能通过抑制与脂质代谢改变相关的抗肿瘤反应来加速卵巢癌的进展。值得一提的是,免疫系统对肿瘤细胞具有监视与清除作用,并且对预防肿瘤发生发展起着重要作用。而内质网应激在免疫系统细胞中的功能已经建立了起来。IRE1α-XBP1信号是无癌宿主中浆细胞、一些树突状细胞群和嗜酸性粒细胞最佳分化所必需的,同时XBP1在巨噬细胞中的表达是Toll样受体激动剂响应产生IL-6所必需的。此外,肿瘤浸润性树突状细胞(tumor-infiltrating dendritic cell,TDC)中XBP1的过度激活扰乱了脂质生物合成过程,并刺激了甘油三酯的异常积累,这一过程与TDC抗原提呈能力降低有关。说明内质网应激反应可以通过阻碍肿瘤微环境中天然免疫细胞的保护功能,削弱抗肿瘤免疫的发展从而促进肿瘤的恶性进展。
3 细胞衰老与癌症
严重或不可逆的细胞损伤会引发脊椎动物细胞周期停滞,与细胞衰老的产生密切相关。衰老是有机体的普遍特征,是由端粒消耗、DNA损伤、氧化应激和某些癌基因激活等多种应激引起的复杂细胞表型。
近年来,关于癌症与细胞衰老之间关系的研究越来越多。一定程度的细胞衰老可能会导致癌症的发生,而在癌症的发生过程中,细胞也可能面临着衰老的威胁。研究表明,细胞衰老可引发增生性的病理变化,在免疫功能低下的小鼠体内注射衰老的人成纤维细胞可显著促进小鼠上皮肿瘤细胞的增殖。这种促增殖作用主要依靠于衰老相关分泌表型(senescenceassociated secretory phenotype,SASP)的组分基质金属蛋白酶3(matrix metalloproteinase 3,MMP3),MMP3还可以促进肿瘤细胞侵袭和血管内皮生长因子(vascular endothelial growth factor,VEGF)形成。除刺激肿瘤细胞增殖外,SASP因子还可诱导上皮细胞间质转化的恶性表型发生。这种形态转变使转化的上皮细胞能够发生迁移和侵袭,推动癌细胞的传播,是原位癌转变为潜在致命侵袭癌的重要步骤。然而,也有研究表明细胞衰老所致的生长停滞也可抑制癌症的发展。衰老可以阻止癌前细胞的繁殖,阻碍癌症的进一步发展。衰老相关分泌表型的组分中的某些因子可以通过自分泌的方式来支持此类生长停滞,例如,在人类黑色素瘤细胞中,IL-6、IL-8和胰岛素样生长因子结合蛋白7(insulin-like growth factor-binding protein 7,IGFBP7)可通过加强大鼠肉瘤病毒癌基因同源突变来诱导细胞发生衰老而引起生长停滞。
另外,在某些特定的条件下,衰老所致的生长停滞是可逆的。这一过程涉及两个主要的抑癌途径p53-p21和p16pRB,这两个途径也被认为是阻止恶性肿瘤发生的屏障。此外,肿瘤细胞中某些癌基因的过表达或抑癌基因的丢失所引发的致癌应激也会导致衰老。综上可知,细胞衰老与癌症相互作用,密不可分。
4 IRE1α通路与细胞衰老在癌症中的相互作用
4.1 IRE1α通路与细胞衰老在癌症中的关系
细胞衰老会引起内质网应激。有研究证实,人类黑色素细胞中致癌基因H-Ras
激活,会导致内质网大量扩张以及引起空泡化相关的细胞衰老表型。在阿霉素诱导的淋巴瘤细胞衰老模型中也报道了内质网超微结构的改变。可见在癌症发展过程中,内质网应激与细胞衰老密切相关。进一步的研究表明,癌基因H-Ras
驱动的衰老是由内质网相关的UPR介导的。但在一定程度上,内质网应激可以延缓衰老。在癌基因依赖模型中,内质网应激的激活通过上调磷酸化AKT和降低磷酸化ERK来减缓衰老。在小鼠和人类黑色素瘤模型中也观察到内质网应激的降低与衰老的增加进一步相关。这种内质网应激瞬时增加导致的衰老延缓也一定意义上促进了肿瘤的发展。可以认为,衰老是激活致癌基因反应的趋同机制,但UPR对其影响则是选择性的,一定程度上UPR有作为肿瘤控制的守门人的直接作用。IRE1α通路作为UPR信号网络的一个重要分支,其与细胞衰老的关系值得进一步探究。研究证明IRE1α通路的激活在细胞衰老中普遍存在。在衰老的WI-38人胚肺细胞中,IRE1α-XBP1s通路被激活,IRE1α激酶和RNase结构域同时被激活,IRE1α蛋白水平明显升高,XBP1s mRNA含量显著增加,细胞活力显著下降,进一步促进细胞衰老表型。在致癌基因H-Ras
所致的黑色素细胞衰老模型和阿霉素诱导DNA损伤所致的淋巴瘤细胞衰老模型中,XBP1s mRNA表达量也明显升高。这种IRE1α-XBP1s通路显著激活的分子依据,与细胞衰老表型相互验证,相辅相成,共同推动肿瘤的发生发展。而当IRE1α信号通路发生阻断时,细胞衰老也相应减缓。在原代小鼠角质细胞中构建H-Ras G
癌基因所致的衰老模型,选择性敲低XBP1
基因后发现,β-半乳糖苷酶(细胞衰老标志物)阳性细胞数量显著减少,这与黑色素细胞衰老模型的结果一致,证实了IRE1α-XBP1s通路在细胞衰老中的加速作用。另有研究表明,IRE1α是早期癌基因Ras
转录后调控的一个重要节点,也可能是Ras
诱导衰老逃逸的重要机制。当内质网应激和IRE1α介导的XBP1s增强H-Ras
诱导的增殖时,IRE1α介导的RIDD则可通过降解原癌因子Id1 mRNA促进早衰。这种既依赖于致癌信号,又依赖于肿瘤微环境所赋予的应激信号,可能是促使其逃避Ras诱导衰老的重要机制,进而也促进了肿瘤的发展。综上可知,细胞衰老常常伴随着IRE1α信号通路的激活,两者相互作用,在癌症发展过程中互为纽带,协调运作。
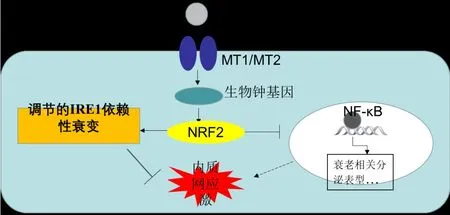
图2 褪黑素通过NRF2-ERAD途径抑制IRE1α-XBP1s通路的激活
4.2 IRE1α通路与细胞衰老在肿瘤细胞中相互作用的机制
4.2.1 NRF2-ERAD途径
目前关于癌症中IRE1α通路与细胞衰老的相互作用机制还不明朗。内质网相关降解ERAD在内质网应激反应中具有识别末端错误折叠蛋白并使它们被降解的功能,核转录因子红系相关因子2(nuclear factor erythroid2-related factor 2,NRF2)作为细胞氧化应激的关键因子,也是ERAD相关基因的主要调节因子。最近的研究指出,NRF2-ERAD途径可能是介导IRE1α通路与细胞衰老相互作用的机制之一。众所周知,褪黑素是一种常见的抗衰老物质,它能够直接去除活性氧类物质。在缓解脂肪间充质干细胞(canine adipose-derived mesenchymal stem cells, cADMSCs)中 , 抗 衰老物质褪黑素可以通过褪黑素受体(melatonin receptor,MT1/MT2)作用于NRF2,通过NRF2诱导的ERAD缓和内质网应激状态,见图2。NRF2对内质网应激进行抑制时减弱了UPR标志物的表达,其中就包括XBP1。这与最近一项研究结果类似,Choi等发现褪黑素可以通过激活ERAD来降低角膜成纤维细胞的内质网应激水平。此外,在NRF2活化剂处理的人皮肤成纤维细胞中,蛋白酶活性增加,衰老也得到有效缓解。因此可以认为,在应对活性氧所致的细胞衰老中,细胞可能通过NRF2-ERAD途径抑制IRE1α-XBP1s通路的激活,使得IRE1α、XBP1s表达下降,细胞存活率提高。4.2.2 细胞凋亡途径
细胞凋亡指细胞为维持内环境稳定而选择的自主有序的死亡。与细胞坏死不同,细胞凋亡是一个主动过程,其目的是为了更好地适应生存环境。当UPR无法缓解时,IRE1α可激活JNK信号通路,介导细胞凋亡。研究表明,衰老可增强内质网应激状态下巨噬细胞的凋亡,并且与IRE1α-XBP1s通路相关,表明凋亡途径可能是介导癌症中IRE1α-XBP1s与细胞衰老相互作用的另一机制。经衣霉素处理后,相比于年轻的巨噬细胞,老化的巨噬细胞出现更加明显的凋亡现象,其磷酸化IRE1α水平明显减少而GRP78蛋白水平显著增加。XBP1
基因选择性敲除可降低经衣霉素处理后老化巨噬细胞的凋亡,证明内质网应激下,老化的巨噬细胞凋亡增强具有IRE1α依赖性。而在小鼠肿瘤模型中,巨噬细胞通过刺激血管生成、增加肿瘤细胞迁移、侵袭和灌注、抑制抗肿瘤免疫等途径促进肿瘤的发生和恶性进展。由此可知,在分子水平上,细胞衰老降低了内质网应激时巨噬细胞内IRE1α的激活,且对XBP1进行抑制可以IRE1α依赖的方式增强巨噬细胞IRE1α的激活并改善其相关凋亡。总的来说,衰老通过这种相互作用增强了内质网应激状态下巨噬细胞的凋亡,进而对肿瘤的进一步发生发展产生影响。5 结语与展望
内质网作为真核细胞特有的管状网络结构,负责蛋白质加工和转运,在钙稳态、蛋白质和脂质合成中起着重要作用。肿瘤细胞增殖迅速,迁移和侵袭也常有发生,特殊的微环境使其长期处于内质网应激状态,UPR持续激活。IRE1α通路可通过XBP1 mRNA的剪切,ERAD和RIDD等途径来支持肿瘤细胞存活,促进癌症的进一步发展。另外,细胞衰老与癌症也密不可分,其与IRE1α-XBP1s通路相互作用,在癌症发展中相辅相成。尽管现有的研究中有很多证据支持UPR与衰老之间存在着紧密的联系,然而,目前对UPR和衰老之间在癌症中的相互作用还有待挖掘,两者相互作用的确切机制也尚不清楚。在未来的研究中,可重点关注UPR特别是IRE1α通路是否通过促进衰老诱导癌细胞的潜伏以及耐药性,以及靶向该UPR通路对逆转衰老及其诱导的耐药性等表型的作用和治疗价值。
