羟基自由基与典型苯系物的后续反应机理研究*
2021-02-07陈笑笑孙延慧
陈笑笑,刘 琳,孙延慧
(青岛科技大学环境与安全工程学院,山东 青岛 266042)
0 引 言
苯系物是苯及其衍生物的总称,包括苯、甲苯和苯乙烯等.苯系物为无色或淡黄色透明油状液体,具有芳香气味,室温下可挥发,温度越高,挥发度越大,且易燃有毒[1].作为有机合成的化学原料,苯系化合物可以通过石化过程挥发产生[2-4],其广泛用于化学生产.张玉欣等[5]解析了南京北郊冬季挥发性有机物的来源,研究表明苯、甲苯、乙苯、间二甲苯、对二甲苯和苯乙烯的含量占总芳香烃的91.64%,其中甲苯含量最高,为总芳香烃的53.40%,苯含量次高,占总芳香烃的17.07%.目前对二甲苯是使用最广泛的苯衍生物,2015年对二甲苯的全球年产量达到了 3.7 亿 t[6].据杨婷等[7]报道,我国城市大气中苯系物的浓度普遍高于其他国家城市;2014年,根据Lim等[8]介绍,世界卫生组织国际癌症研究机构(IARC)将苯列为一类致癌物,甲苯和二甲苯均为三类致癌物.苯系物不仅对人类健康产生直接的影响,还能够引起城市的光化学烟雾,产生的二次有机气溶胶等对人类健康有更大的危害.
大气中的苯系物主要通过与羟基自由基(·OH)、硝基自由基(·NO3)和臭氧分子(O3)反应,其中·OH是大气中非常重要的氧化剂[9-10].在(295±2)K 和95 kPa的合成空气或氧气(O2)流量下,Berndt和Boge[11]研究了·OH与苯的气相反应.在不含氮氧化物(NOx)的情况下,用长程傅里叶变换红外谱仪(FTIR)和紫外吸收光谱(UV)法在多种实验条件下测定了·OH与苯反应生成苯酚的量,同时检测到了乙二醛、顺丁烯醛和反丁烯醛.在NOx存在时,检测到的羰基化合物为乙二醛、顺丁烯和反丁烯,也有微量的硝基苯和邻硝基苯酚.Olivella等[12]通过密度泛函理论(density functional theory,DFT)和量子力学研究了苯酚自由基(苯-OH)与O2的反应途径,O2加成到苯-OH上所产生的过氧自由基的反式立体异构体比顺式更稳定;Uc等[13]通过量子化学方法和常规过渡态理论研究了200~600 K温度范围内·OH引起的苯和甲苯的加成与氢(H)原子抽提反应,得到了相应的速率常数,但未考虑·OH抽提甲苯中苯环 H原子的反应过程;Fan和 Zhang[14]阐明了·OH引发的对二甲苯大气氧化的反应途径,预计·OH主要添加在邻位,计算的·OH与对二甲苯的反应速率常数与实验研究一致,但并未研究·OH抽提对二甲苯甲基H原子的反应途径.到目前为止,·OH与甲苯、对二甲苯的反应机理仍需要补充,而苯-OH在O2、·NO3的浓度下的产物已经进行了多次实验研究,但并未全面地考虑其在一般的大气条件下受到多种大气氧化剂的影响,如何自发地进行热力学反应.
近年来,DFT为预测反应过程产生的自由基中间体,监测反应路径和阐明最终产物提供了理论基础,已广泛应用于大气环境领域.DFT常用方法包括 B3LYP、M06泛 函 、CCSD(T)和 MPWB1K 等.B3LYP方法是具有较好的低成本性能,但与其他混合密度函数相比,B3LYP在预测电子能量时,准确性较差[15];M06泛函适合计算具有分子间弱相互作用的体系[16],其在平衡阴离子系统的计算速度和精度时,提供了一个相当可靠的选择,但通常会高估芳香族配位化合物(静电力)的相互作用能[17];CCSD(T)方法相对比较准确,但会耗费非常大的计算成本;MPWB1K是计算屏障高度和反应能量的最佳DFT 方法之一[18],与其他 DFT 计算方法相比,MPWB1K具有更好的叠加效应,通过MPWB1K功能的一系列研究取得了令人鼓舞的成果[19-20].DFT/MPWB1K在生物大分子的研究中具有很好的应用前景,或者至少可以用于分子力学势的参数化和其他计算技术[21].
为了深入理解苯系物的大气反应机理,进一步完善其在大气中的清除、迁移和转化机制.本文采用量子化学计算方法,研究了·OH与苯、甲苯和对二甲苯的反应机理及相应的热力学性质(能垒和反应热),系统地阐述了·OH与苯的产物在大气中可能会经历的一系列后续的化学变化.同时,本文也验证了MPWB1K方法计算结果的准确性,基于 MPWB1K/6-31+G(d,p)优化完几何结构后,在 MPWB1K/6-311+G(3df,2p)和 CCSD(T)/6-311+G(d,p)的理论水平分别计算·OH氧化降解苯与甲苯的反应能垒(ΔE)和反应热(ΔH),并将二者进行比较.期望本文可以补充实验结果,并为其他苯系物的研究提供新的研究思路.
1 计算方法
本文所有量子化学计算都是通过Gaussian 09软件包(Revision,A.02)进行的.采用 MPWB1K 方法,使用 6-31+G(d,p)基组对反应物、中间体、过渡态和产物的几何结构进行了全面优化.在结构优化的基础上,确定了相同水平下的谐波振动频率,以确定静止点的特性,即零点能(ZPE).在计算过程中,反应物和中间体没有虚频,过渡态有且只有1个虚频率.内禀反应坐标(IRC)理论用于确定指定反应物和预期产物之间的过渡结构[22].基于优化的结构,使用更精确的基组 6-311+G(3df,2p)[23],用以计算反应物、中间体、过渡态和产物的单点能量和总电子能量.
2 反应机理
在大气条件下,对于苯、甲苯和对二甲苯与·OH的反应来说,主要有·OH加成和H原子抽提2种可能的反应方式.IM为反应过程生成的中间体,TS为反应过程中的过渡态.
2.1 ·OH与苯的反应机理
由于苯环是高度对称的,苯环上的6个碳(C)原子完全相同,因此,只考虑1个·OH加成反应和1个H原子抽提反应.相应的反应能垒和反应热如图1所示.与H原子抽提途径相比,OH的加成反应具有更低的能量,该反应生成的中间体苯酚自由基(IM1)更稳定,其在热力学上也更有利.但中间体IM1和IM2在大气中仍不稳定,可以被进一步氧化.

图1 ·OH与苯的反应机理
2.2 ·OH与甲苯的反应机理
甲苯具有对称性,4个可能的·OH加成位置分别是 C1、C2、C3和 C4,反应机理如图 2所示.比较所有的·OH加成通道,C2位置的·OH加成反应能垒最低且放热最多,最容易发生反应.研究表明:甲基(—CH3)基团产生的诱导效应会引起C2原子的电荷密度增加,利于·OH与C2的加成.甲苯苯环上有4个不同的C原子,但C1上没有H原子,因此,只考虑苯环C原子(C2、C3和C4)上的H和—CH3基团中C原子上的H,这4种不同的H对应4种不同H原子抽取.如图2所示,反应过程中的TS3是·OH抽提—CH3上的H,最低ΔE=0.50 kcal/mol,ΔH=-26.97 kcal/mol,得到产物IM3和水(H2O).
2.3 ·OH与对二甲苯的反应机理

图2 ·OH与甲苯反应机理
对二甲苯具有对称性,苯环上有2种不同的C原子,因此,有2种不同的·OH反应通道.对二甲苯的这2步·OH加成路径的ΔE较低,均≤1.00 kcal/mol,在大气条件下很容易发生.类似地,对二甲苯有2种不同的H原子,即苯环上C2连接的H和—CH3上的H.通过比较这2个H抽提通道,可以得出·OH提取甲基上的H更容易发生,得到产物IM11和H2O.

图3 ·OH与对二甲苯反应机理
3 后续反应机理
大气环境中含有丰富的活性自由基和分子,如一氧化氮(NO)、二氧化氮(NO2)、O2和H2O等.苯、甲苯和对二甲苯初级反应产生的活性中间体将在大气中被进一步氧化生成羟基或硝基化合物,下面以苯产生的IM1和IM2为例,详细讨论其在大气中的后续反应路径.
3.1 IM1的后续反应
本文考虑了IM1在大气中可能发生的6种后续反应路径,包括H原子脱去反应,H原子抽提反应,以及与O2和NO2的加成反应,反应机理如图4所示.IM1可以直接脱去H原子,也可以通过与O2反应去除过氧化氢自由基(·HO2)的方式抽提H原子,得到相同的产物苯酚.反应路径的势能剖面图如图5所示:第1步H抽取反应需要跨越23.26 kcal/mol的能量屏障,并且ΔH=14.03 kcal/mol;第 2 步加 O2去除·HO2的ΔE=6.60 kcal/mol,ΔH=-31.74 kcal/mol.可以得出,通过过渡态TS1b形成苯酚的过程能垒较低并且放热较多,在热力学中更具有优势,是大气条件下产生苯酚的主要途径.
对于IM1与O2的直接加成反应,考虑到·OH的空间位阻效应,主要有2种不同的反应模式.O2分子可以从OH的同侧或异侧加成到苯环上,即顺式和反式加成.这2步加成反应的ΔE分别为10.20和8.41 kcal/mol,均是放热反应,ΔH分别为-8.22和-6.68 kcal/mol,在大气中比较容易发生.IM1c和IM1d中间体都具有较高的反应活性,易于在大气中被进一步氧化.考虑到IM1c和IM1d具有类似的结构,选择能容易产生的IM1d进行详细的后续研究.IM1与NO2的反应与IM1与O2的反应类似.具有2种不同的NO2加成方式,顺式和反式加成.这2步NO2加成均是无垒的,且放热较多,ΔH分别为-35.71和-37.94 kcal/mol.得到2个IM1e和IM1f活性中间体,可以通过小分子脱除反应得到产物邻硝基苯酚和硝基苯,但是由于需要克服较大的能量屏障(ΔE分别为75.63和43.82 kcal/mol),这2条路径极难实现.
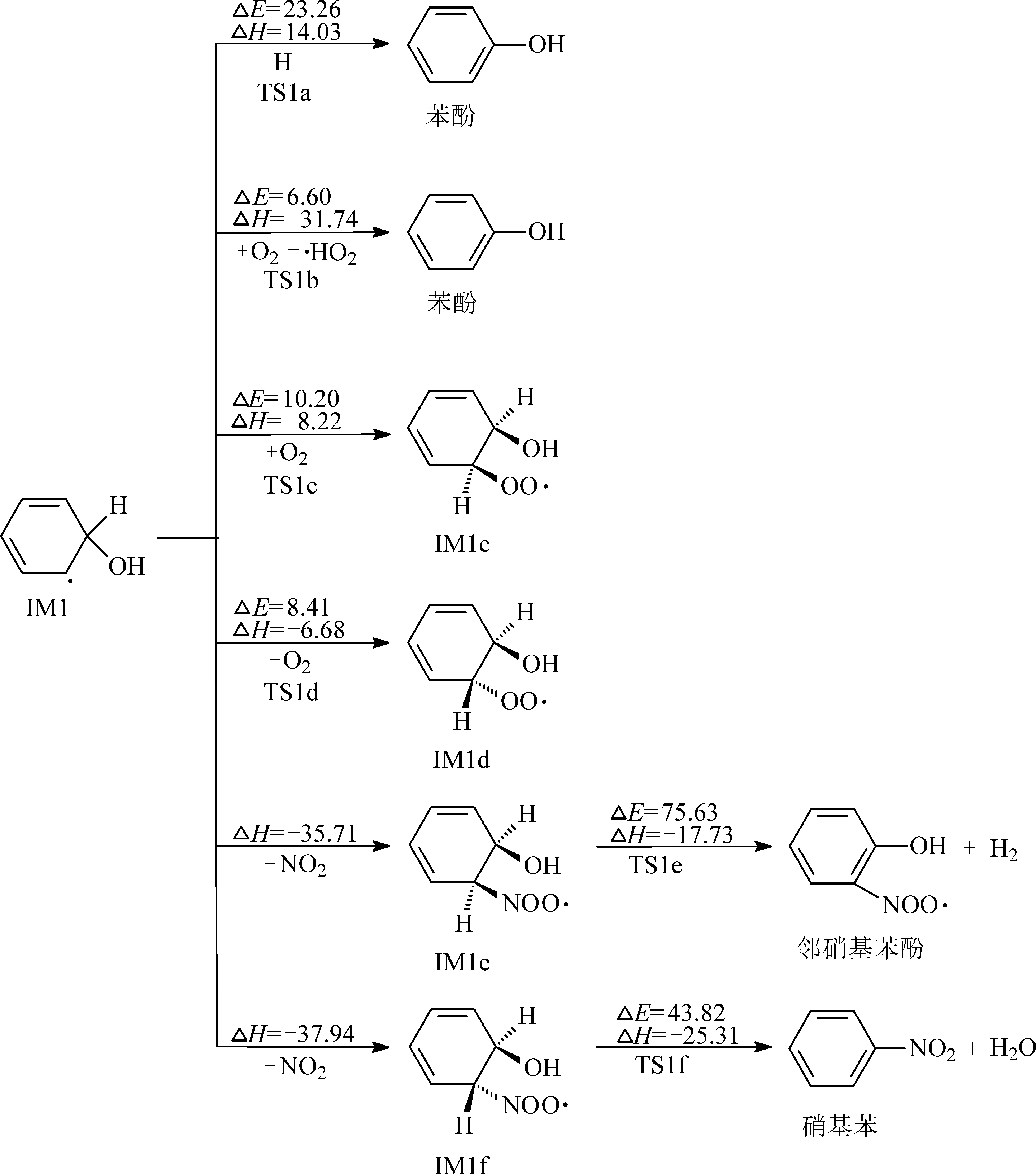
图4 IM1的后续反应路径

图5 IM1后续反应路径的势能剖面
3.2 IM1d的后续反应
IM1d活性中间体可以在大气中进一步反应.为了更好地揭示IM1d的大气降解机理,本文主要研究了IM1d中H迁移反应,与NO的反应以及自身异构化反应.
3.2.1 H迁移反应
IM1d中羟基(—OH)基团上的H原子与过氧(—OO)基团空间距离较近,可以转移到—OO基团,形成过氧化氢(—OOH)基团,ΔE=24.14 kcal/mol,生成 IM1d1,ΔH=-17.82 kcal/mol.然后 IM1d1脱去1个 ·HO2,通过过渡态 TS1d2得到苯酚,ΔE=10.01 kcal/mol,ΔH=-7.24 kcal/mol,迁移机理如图 6所示.在IM1d中,H迁移过程具有相对中等的能量障碍,基于热力学数值分析,该途径是可以发生的.
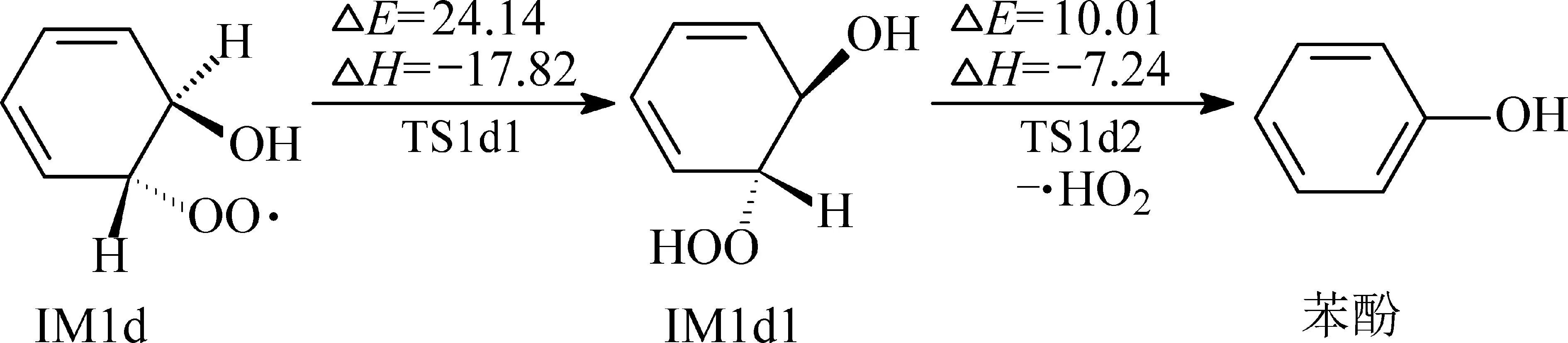
图6 IM1d中H的迁移机理
3.2.2 与NO反应
IM1d与NO的加成是无垒的,类似于IM1与NO2的加成过程,其反应机理如图7所示.NO分子可以直接加成到IM1d的—OO基团末端的O原子上,形成 IM1d2,ΔH=-15.13 kcal/mol.IM1d2中间体是不稳定的,可以发生单分子分解,脱去NO2,形成IM1d3活性中间体,该过程需要克服反应势能(ΔE=20.80 kcal/mol),并且释放能量(ΔH=-1.74 kcal/mol).随后,IM1d3 经历开环,与大气中O2反应,最终得到1个二醛化合物.IM1d与NO的加成及后续反应,能垒较低且都是放热的,在热力学上比较容易发生.
3.2.3 自身异构化反应

图7 IM1d与NO反应机理
IM1d异构化反应如图8所示:IM1d可以自身异构化发生闭环反应,过氧自由基(·O2)上的末端O原子可以攻击苯环上的C原子,形成桥环化合物IM1d5,该过程的ΔE=18.10 kcal/mol,ΔH=-7.39 kcal/mol;IMld5在大气中可以进一步与O2结合,产生过氧桥环化合物 IM1d6,ΔE=15.96 kcal/mol,ΔH=-12.93 kcal/mol;IM1d6可以与NO加成生成IM1d7,释放热量(ΔH=-16.18 kcal/mol);IM1d7 进而发生 NO2脱除,得到含氧桥环化合物IM1d8,中间体IM1d8是不稳定的,在大气中经历一系列反应,包括开环、异构化和断键等,最终得到产物乙二醛和顺丁烯二醛.

图8 IM1d异构化反应
3.3 IM2的后续反应
IM2的后续反应路径和势能剖面图如图9和10所示.IM2具有3种不同的反应,分别与O2、·OH和NO2加成.这3种加成反应都是无垒的,释放热量较多,ΔH分别为-45.82、-110.04和-73.12 kcal/mol.与O2加成得到的IM2a过氧加合物可以继续与NO反应生成 IM2a1,该过程的ΔH=-15.52 kcal/mol;随后,IM2a1中O—O键破裂,单分子分解产生含氧自由基IM2a2和NO2;最后,IM2a2可以抽提H2O中的H原子形成苯酚.
4 方法验证
为了验证 MPWB1K/6-31+G(d,p)方法优化几何结构的准确性,本文将苯酚、水和苯的几何参数,包括化学键键长和键角,与计算化学比较和基准数据库(computational chemistry comparison and benchmark database,CCCBDB)[24]提供参考的实验数值进行比较,详细结果如表1所示.通过比较可知,苯酚、H2O和苯的化学键键长的相对误差分别为-0.89%~0.28%、-0.52%和-0.87%~-0.46%,绝对值均<1.00%;对应键角的相对误差分别为1.09%、1.60%和0.因此,使用MPWB1K方法优化结构能够得到相对合理的几何构型.
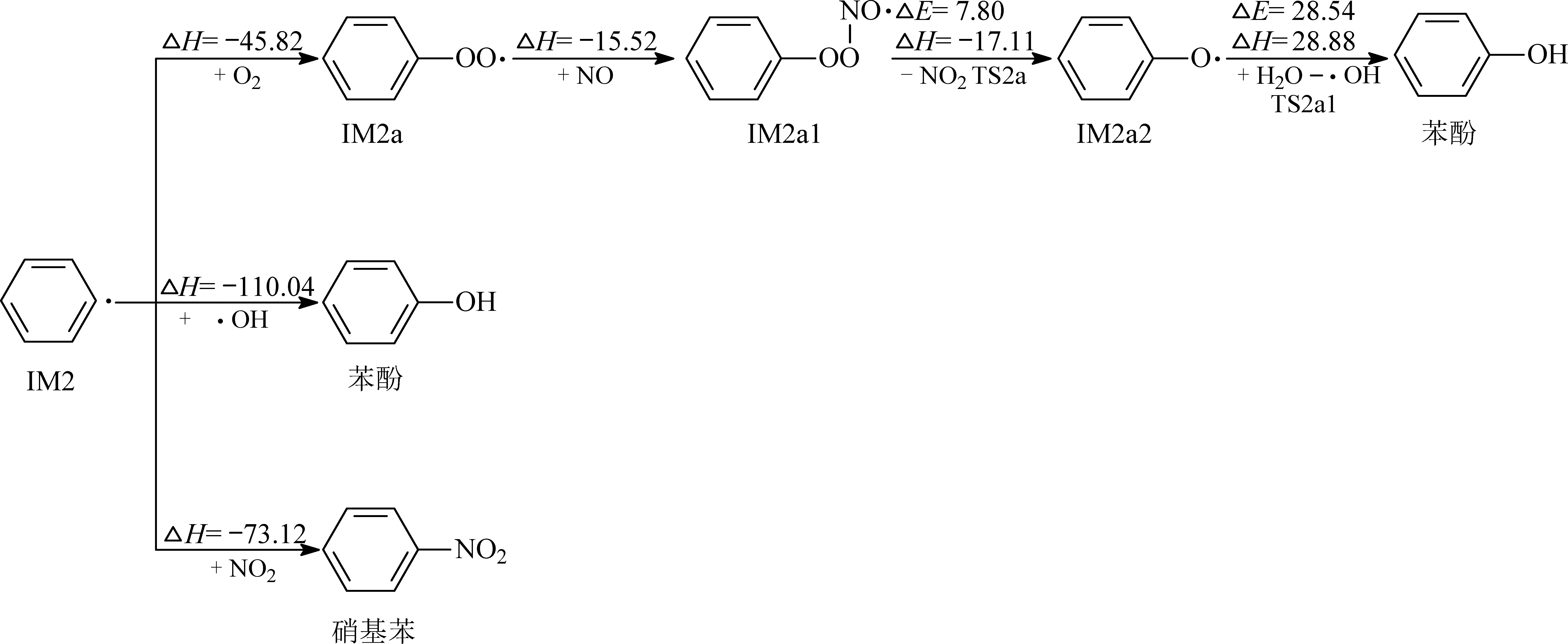
图9 IM2的后续反应路径
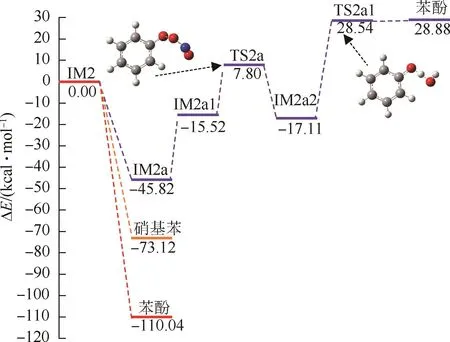
图10 IM2后续反应路径的势能剖面
4.1 ·OH与苯反应机理的方法比较
在使用 CCSD(T)/6-311+G(d,p)方法计算·OH与苯反应机理的单点能量时,加成过程的ΔE=3.75 kcal/mol,ΔH=-11.68 kcal/mol;H原子抽提过程中的ΔE和ΔH分别为7.06和-2.21 kcal/mol.与MPWB1K/6-311+G(3df,2p)理论水平计算的数值比较,二者之间最多相差2.51 kcal/mol,并且加成过程都比H原子抽提过程更有利.然而CCSD(T)/6-311+G(d,p)方法所占用的 CPU 时间为 1 h 8 min 24.7 s~10 h 44 min 43.6 s,远远高于后者花费的时间(14 min 34.0 s~29 min 28.2 s).
4.2 ·OH与甲苯反应机理的方法比较
在 CCSD(T)/6-311+G(d,p)//MPWB1K/6-31+G(d,p)与 MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p)理论水平下,·OH与甲苯反应的ΔE和ΔH如表2所示.只有甲苯+·OH→IM3+H2O的相对能量差为3.70 kcal/mol,其余反应路径相对能量差均<2.80 kcal/mol.基于这2种方法的计算结果,ΔE和ΔH均显示出合理的差异,最优反应路径具有良好的一致性.但是CCSD(T)方法占用工作站的CPU时间为3 h 14 min 5.2 s~1 d 9 h 32 min 33.8 s,然而使用后者时均≤1 h 27 min 11.8 s.因此,基于计算精度和效率,MPWB1K理论更适用于描述反应系统的能量计算.

表1 苯酚、水和苯的几何参数计算结果与参考值比较

表2 不同理论水平下计算的·OH与甲苯反应的能量比较 单位:kcal/mol
5 结束语
本文采用MPWB1K方法,使用 6-31+G(d,p)基组对·OH与苯、甲苯和对二甲苯的初始反应中涉及的所有物质进行了结构优化,并运用6-311+G(3df,2p)基组计算了相应的单点能.其中·OH与苯的H原子抽提反应的ΔH=-4.15 kcal/mol,略低于Lin等[25]实验报道的 5.09 kcal/mol;Suh 等[26]提到实验测量的甲苯-OH加成物的4种异构体的ΔH=(-16.50±5.00)kcal/mol,并且最优路径是邻位加成,本文计算的数据很好地符合实验测量结果.·OH主要加成在对二甲苯的邻位,这与Fan和Zhang[14]的计算结果一致.本文对·OH与苯随后的反应进行了更全面更具体的理论研究,并预测了苯酚、硝基苯、乙二醛和顺丁烯二醛等物质的生成,与 Berndt和 Boge[11]的实验报道的一致.
本文详细研究了苯、甲苯和对二甲苯与·OH的加成和H原子抽提反应过程,补充了先前研究的不足.热力学计算表明,·OH与苯、甲苯及对二甲苯的加成反应均比H原子抽提反应更容易发生.另外,·OH与苯的反应产物在大气中如何进行后续的自发反应也被考虑.首先选取苯与·OH产生的IM1活性中间体研究其后续反应,其中IM1与O2的反应最易发生,主要产生IM1d过氧化合物.IM1d可经历3种反应通道:H迁移反应、与NO反应以及自身异构化反应,最终生成产物乙二醛、苯酚和硝基苯等.苯与·OH产生的IM2抽提中间体在大气中也会进一步与O2、·OH和NO2反应,最终生成苯酚和硝基苯.产物乙二醛和苯酚等具有很高的反应活性,在大气中会被进一步氧化,最终生成二次有机气溶胶.
