中国东部水稻土壤丁酸互营降解微生物的地理分布格局
2021-02-02费媛媛焦硕陆雅海
费媛媛 焦硕 陆雅海
中国东部水稻土壤丁酸互营降解微生物的地理分布格局
费媛媛 焦硕 陆雅海†
北京大学城市与环境学院, 北京 100871; †通信作者, E-mail: luyh@pku.edu.cn
沿纬度梯度收集中国东部 34 个水稻土壤样品, 在实验室条件下, 以丁酸钠为底物, 进行二次厌氧富集实验。运用微生物高通量测序技术, 分析样地水稻土壤中丁酸互营降解过程的微生物群落特征、功能活性及互营单胞菌相对丰度地理分布格局。结果表明, 丁酸降解产甲烷的延滞期(3~14 天)随着样地纬度的升高而加长, 但最大产甲烷速率没有显著的差异。环境因子相关分析结果表明, 丁酸降解细菌的关键类群为互营单胞菌(), 互营单胞菌的相对丰度主要受年平均温度(MAT)的影响。互营单胞菌相对丰度较大的样地, 丁酸完全降解所需时间较短。丁酸互营降解微生物的群落结构表现出显著的距离衰减关系(<0.05), 表明微生物群落的构建同时受到空间距离和环境因子的驱动。
丁酸降解; 产甲烷; 互营单胞菌相对丰度; 微生物地理分布
甲烷(CH4)气体的排放源较为复杂, 大致分为自然排放源和人为排放源。农业排放源约占人为排放源的 14%以上[1], 其中水稻土壤是主要的农业排放源之一。水稻土壤中有机物厌氧降解产生甲烷的过程主要分为 3 个阶段。首先, 植物残体等有机质大分子在初级发酵菌作用下分解成小分子物质(如脂肪酸和醇类等); 然后, 在次级发酵菌(又称互营细菌)的作用下, 小分子物质进一步分解成乙酸、H2和 CO2等甲烷前体物质; 最后, 在产甲烷菌的作用下, 甲烷前体物质转化成 CH4[2–3]。最复杂的环节为短链脂肪酸的厌氧降解, 丁酸是有机物降解的重要中间产物之一[4]。本文将丁酸通过厌氧降解产生CH4的过程简称为丁酸互营降解。
在热力学条件限制下, 丁酸互营降解过程是互营细菌和产甲烷古菌共同作用的结果[2], 其中的互营细菌主要为互营单胞菌科 Syntrophomonadaceae。Zou 等[5]的研究表明, 水稻土壤中参与丁酸互营降解的产甲烷古菌主要为甲烷八叠球菌科(Methano-sarcinaceae)和甲烷胞菌目(Methanocellales)。通常认为, H2和甲酸盐是丁酸互营氧化过程中的主要电子载体, 但我们在前期的研究中发现导电特性纳米材料(如纳米磁铁矿和碳纳米管)可以显著地促进丁酸互营氧化, 推测可能存在直接种间电子传递作用(direct interspecies electron transfer, DIET)[6]。水稻土壤和湖泊底泥等缺氧环境中参与丁酸盐降解和CH4生成的互营细菌主要为互营单胞菌和地杆菌(), 产甲烷古菌为甲烷八叠球菌、甲烷胞菌和甲烷杆菌(s)等[7–9]。
研究微生物群落的地理分布格局, 旨在探索它们的栖息地、数量和多样性等特征及机理[10]。距离衰减关系(distance−decay relationship, DDR)常用于表征微生物的生物地理模式, 表示微生物群落相似性随着地理距离的增加而降低, 体现群落的多样性在空间尺度上的变化[11]。一般来说, 对微生物群落产生影响的环境因子主要有温度、pH、盐度和地理距离等[12]。另外, 在丁酸互营降解过程中, 丁酸盐氧化菌受到热力学条件限制[2], 因此不但难以被分离培养, 而且在土壤微生物群落中占比很小, 使得原位研究的难度较大。
本文从我国东部主要稻米生产区采集 34 个水稻土壤样品, 通过实验室培养和分子生态学技术相结合的方法, 研究水稻土壤丁酸互营降解过程及关键微生物种类的地理分布特征, 旨在为预测不同地区水稻土壤中有机质厌氧分解、丁酸等脂肪酸中间产物的动态变化以及甲烷排放的空间变异特征提供科学依据。研究内容包括: 1)通过观察不同地点水稻土壤的产甲烷延滞期、最大产甲烷速率及丁酸完全降解时间, 探究丁酸互营降解过程的区域地理特征; 2)探究丁酸互营降解微生物的地理分布格局及环境因子的影响。
1 材料与方法
1.1 土壤样品采集和分析
于水稻种植季节(2017 年 7—9 月), 沿纬度梯度自北向南, 从我国东部稻米主要生产区(108.8885°—126.9806°E, 18.4466—46.6135°N)采集 34 个耕作层样品。在每个样地随机选取 3 个采样点(每个采样点的面积均为 100m2), 依据五点采样法, 在每个采样点对角线的中点及外围四处(与中点距离相等), 在 0~15cm 深度范围内采取 5 个土壤样本, 保证每个土壤样本的采样深度及采样量均匀。先将每个采样点的 5 个样品混合, 再将 3 个采样点的土壤样本混合, 成为该样地最终的土壤样本。将样本运回实验室, 置于 4℃冰箱内保存[13]。一部分样品在室温下自然风干, 用 2.0mm 筛子去除植物根系和石头等残留物, 用于土壤理化性质的分析。
用标准土壤试验方法[14]分析土壤样本的物理化学特性, 包括 pH、阳离子交换量(CEC)、有机质含量(OM)、溶解性有机碳(DOC)、微生物生物量碳(MBC)、微生物生物量氮(MBN)、硝态氮(NO3−)和铵态氮(NH4+)、总氮(TN)和有效氮(AN)、总磷(TP)和有效磷(AP)、总钾(TK)和有效钾(AK)、总硫(TS)和有效硫(AS)、总锰(TMn)和有效锰(AMn)、总铜(TCu)和有效铜(ACu)、总铁(TFe)和有效铁(AFe)、总锌(TZn)和有效锌(AZn)。此外, 从 WorldClim 数据库(https://www.worldclim.org/)获得 34 个样地的气候数据, 包括年平均温度(mean annual tempera- ture, MAT)和年平均降水量(mean annual precipita-tion, MAP)[13]。
1.2 丁酸互营降解厌氧培养实验
取适量土壤样本于烧杯中, 按照水土比 5:1 (质量/体积)制备土壤悬浮液, 每个土壤样本设置 3 个平行实验。首先, 将搅拌均匀的土壤悬浮液置于100mL 西林瓶中, 充入纯度为 99.999%的 N2, 置换瓶内气体, 气体置换完毕后, 给每个培养瓶塞上丁基胶塞, 盖上铝盖, 并用封口器封口。然后, 向每个培养瓶中注入 5mmol 的丁酸钠溶液, 再次充入纯N2进行气体置换, 之后将培养瓶放置于 30℃恒温箱中, 开始第一次富集培养实验。土壤悬浮液中的丁酸钠被完全降解后, 再次向培养瓶中注入 5mmol丁酸钠溶液, 气体置换完毕后放置于 30℃恒温箱中, 进行第二次富集培养实验。
将 5mmol 丁酸开始降解至其消耗结束所需时间定义为富集培养时间, 不同样地的培养时间各不相同, 第一代培养时间为 11~22 天, 第二代培养时间比第一代短, 为 4~10天。在富集培养前期, 为了更精确地测定其产甲烷延滞期, 平均间隔 1 天测定一次 CH4浓度。在富集培养中后期, 平均间隔 2 天测定一次 CH4浓度。每一代富集培养丁酸浓度的测定次数为 4~5 次。
本研究对丁酸互营降解过程的特征设置 3 个分析指标: 1)产甲烷延滞期, 即从实验开始到 CH4浓度呈线性增加的时间[10]; 2)利用产甲烷曲线的斜率计算第二代最大产甲烷速率(mmol/d); 3)第二代丁酸完全降解所需时间。
1.3 微生物群落分析和统计方法
1.3.1 16S rRNA 高通量测序分析
富集培养实验结束后, 利用土壤 DNA 快速提取试剂盒(Fast DNA®Spin Kit for Soil, MP, 美国)提取土壤样本的 DNA, 操作步骤按试剂盒说明书进行。细菌的引物为 338F: 5’-ACTCCTACGGGAGG AGCA-3’; 806R: 5’- GGACTACHVGGGTWTCTAA T-3’[15]。古菌的引物为1106F: 5’-TTWAGTCAGGC AACGAGC-3’; 1378R: 5’-TGTGCAAGGAGCAGG GAC-3’[16]。微生物群落的高通量测序由广东美格基因科技有限公司完成, 在Illumina Hi-Seq 2500 平台(Illumina Inc, San Diego, 美国)进行分析。利用USEARCH (v8.0.1517, http://www.drive5.com/usear ch/)进行序列分析, 同时使用UPARSE pipeline, 将相似性≥97%的序列定义为相同 OTU (operational taxonomic unit)[17], 之后筛选出每个 OTU 的代表性序列进行系统发育分类和统计分析。
1.3.2统计分析
采用一元线性回归法, 分析丁酸完全降解时间与互营单胞菌()相对丰度之间的关系。在丁酸完全降解时间与互营单胞菌相对丰度的散点图上添加趋势线, 获得二者的回归方程。通过DDRs 来衡量微生物群落的扩散限制情况。利用 R v3.5.1 语言软件(http://www.r-project.org/), 基于微生物群落的 Bray-Curtis 距离和样地间地理距离两项指标, 对 DDRs 进行分析[18]。通过冗余分析(redun-dancy analysis, RDA)和主坐标约束分析(canonical analysis of principal coordinates, CAP), 分别探讨微生物地理分布格局和环境因素的影响。利用 ordi-R2step 函数选出有显著影响的环境因子(<0.05)[19], 最后进行绘图。
利用 R v3.5.1 中的“maps”包绘制样地地图[20]。利用 R 软件“vegan”包里的“reg”函数进行线性回归计算, 获得互营单胞菌相对丰度与环境因子之间的关系。
为了探索微生物之间的共现性模式, 我们利用“Igraph”包[21]和 The Open Graph Viz 平台[22]进行共发生网络分析。在共发生网络图中, 每一个节点表示一个菌属。在相关系数>0.6,<0.01 的情况下, 认为共发生事件是有效的[23]。共发生网络参数主要包括平均度、节点数、边数、图密度和模块化指数等。
2 研究结果
2.1 丁酸互营降解过程的特征及微生物群落的组成
通过高通量测序和抽平处理后, 对细菌共测得约 13200 个 OTUs, 并划分为 80 个科, 其中最常见的 OTUs (科水平)为 Anaerolineaceae (6.1%), Syntro-phomonadaceae(5.1%)和 Xanthobacteraceae (2.4%), 相对丰度最高的菌属(属水平)为(5.1%)。对古菌测得约 1004 个 OTUs, 划分为 13 个科, OTUs (门水平)分别为 Euryarchaeota(84.8%)和Thaumarchaeota (12.8%), 相对丰度最高的菌属(属水平)为(21.5%)。
丁酸互营降解过程具有以下特征: 1) 产甲烷延滞期随着纬度升高而加长(图 1); 2)相对丰度较大的样地, 丁酸完全降解所需时间较短(图 2)。
2.2 丁酸互营降解微生物的地理分布格局
丁酸互营降解微生物群落表现出显著的距离衰减关系(斜率=−0.011)(图 3), 表明微生物群落的构建同时受到空间距离和环境因子的驱动。为了进一步探究环境因子对微生物群落的驱动作用, 基于微生物群落的 Bray-Curtis 距离进行 CAP 分析, 结果如图 4 所示。环境因子都具有显著影响(<0.05), 温度(MAT)和土壤 pH 对微生物群落产生重要的影响(图4(a1)和(b1))。通过 RDA 分析, 进一步探究不同环境因子对优势微生物(属水平)的影响, 结果表明, 对于细菌群落,和受MAT 和土壤 NH4+的影响最大(图 4 (a2)); 对于古菌群落, 产甲烷菌(,等)和一些其他古菌(_,_等)受 pH 和 AN的影响较大(图 4(b2))。
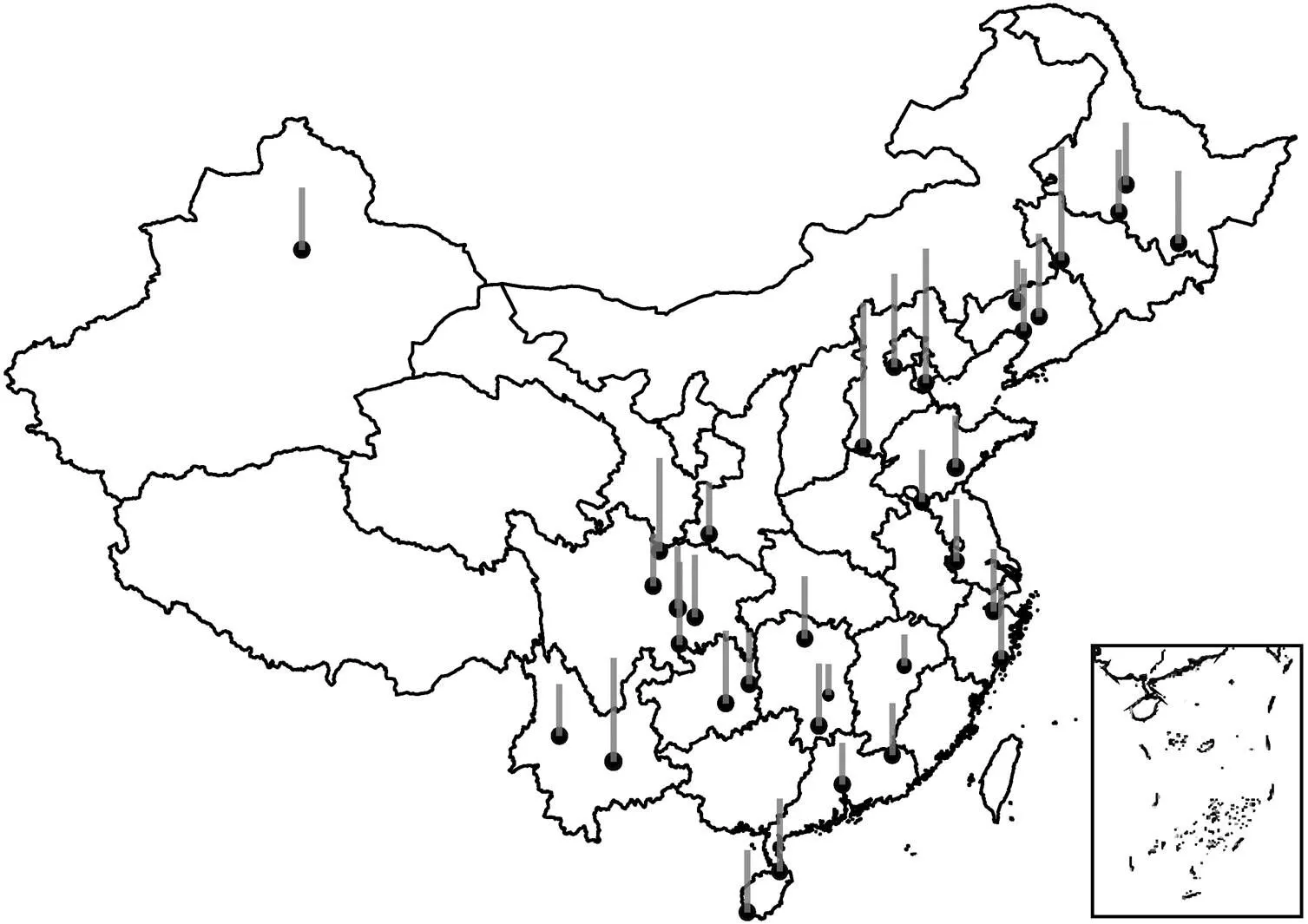
灰色柱子表示延滞期的相对值, 柱子越高, 其值越大
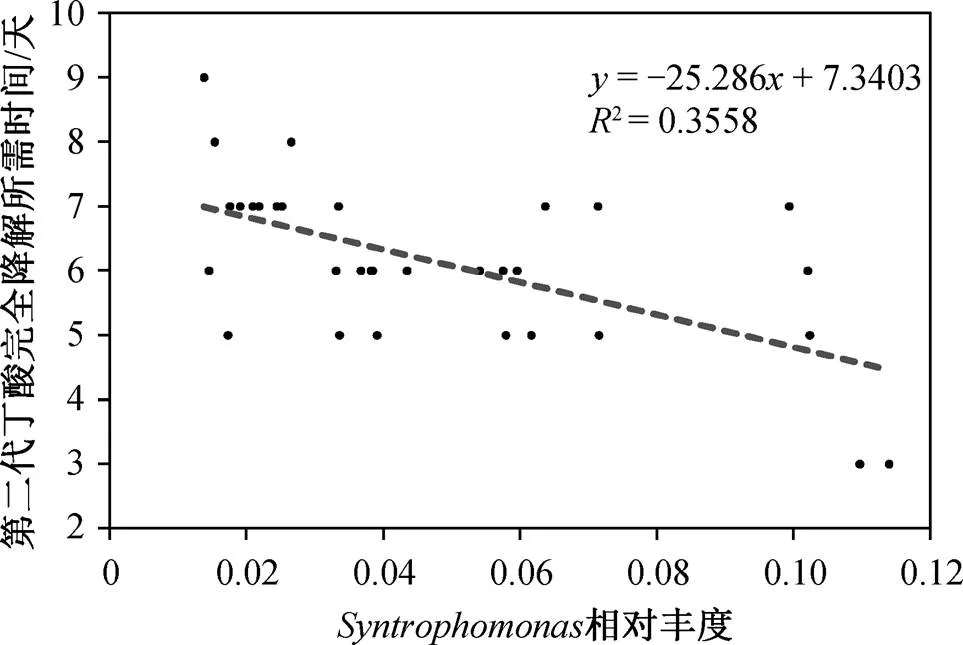
图2 丁酸完全降解时间与菌属互营单胞菌(Syntropho-monas)相对丰度的关系
2.3 丁酸互营降解微生物对产甲烷延滞期和最大产甲烷速率的影响
产甲烷延滞期和最大产甲烷速率取决于微生物群落的结构和功能。Spearman 相关性分析结果表明, 对于古菌群落(图 5(a)),,,,和_的相对丰度均与产甲烷延滞期负相关, 即菌属的相对丰度越大, 产甲烷延滞期越短, 但和_的相对丰度与产甲烷延滞期呈正相关。,,_和的相对丰度均与最大产甲烷速率正相关, 其中与最大产甲烷速率显著正相关(= 0.42,<0.05)。
对于细菌群落(图5(b)),,,,和的相对丰度均与产甲烷延滞期正相关, 但,和的相对丰度与产甲烷延滞期负相关。此外,的相对丰度与最大产甲烷速率显著正相关(= 0.27,<0.05)。
2.4 互营单胞菌作为丁酸降解的关键类群
丁酸互营降解过程中,对丁酸的降解起关键作用, 在细菌群落中其相对丰度最大。图6展现的地理分布格局, 可以看到南部样地中的相对丰度明显大于北部样地。为了进一步探究这种地理分布格局产生的原因, 本文利用线性回归方法分析环境因子对相对丰度的影响。图7显示, 温度(MAT)和NH4+对的相对丰度产生显著的正面影响(<0.05)。

图3 水稻土壤丁酸互营降解微生物群落的距离衰减关系
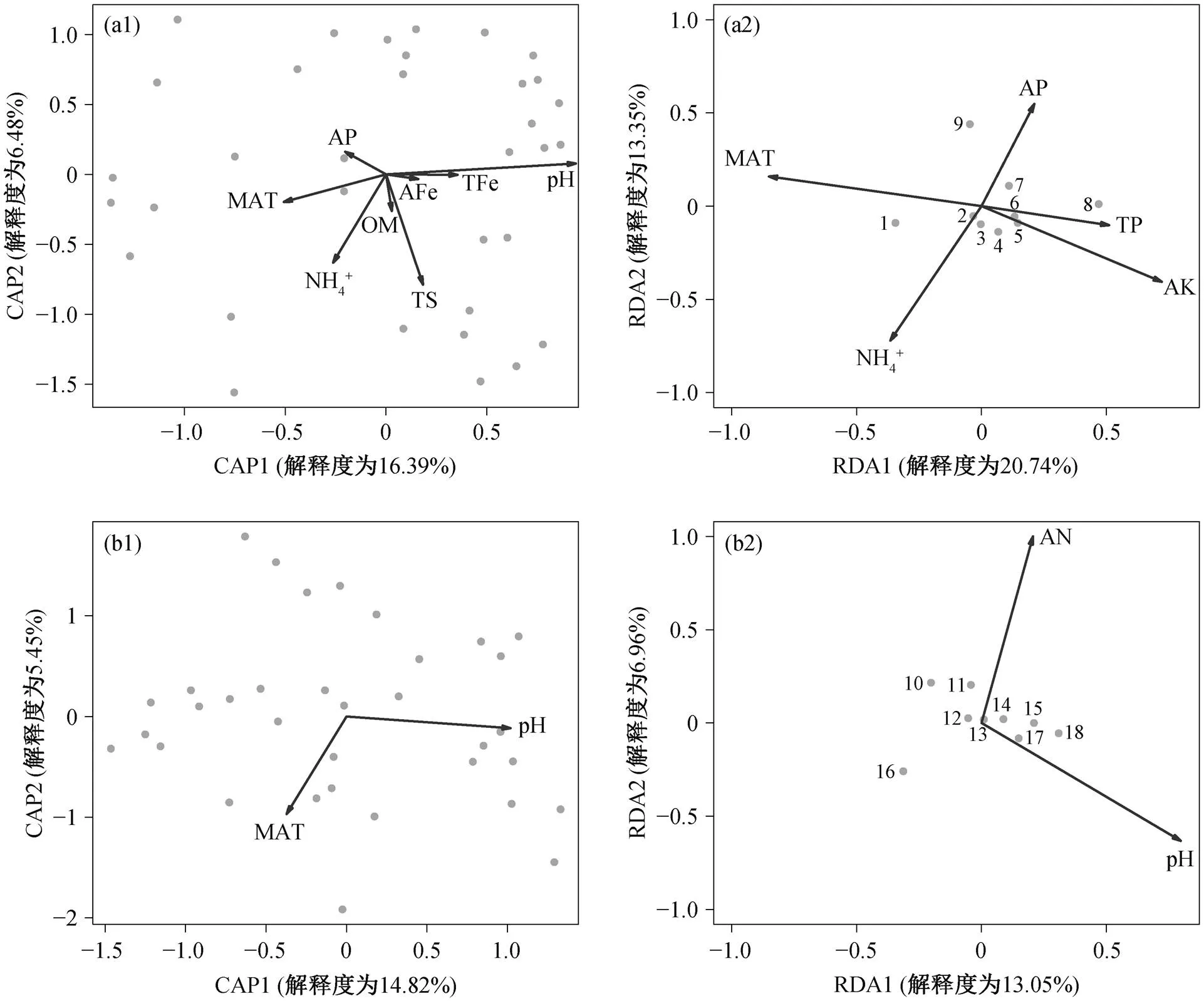
(a1)和(a2)细菌; (b1)和(b2)古菌。1.Syntrophomonas; 2.Geobacter; 3.Anaeromyxobacter; 4.ADurb.Bin063.1; 5.Anaerolinea; 6.Candidatus_Solibacter; 7.Gaiella; 8.Pseudarthrobacter; 9.Bacillus; 10.Methanoregula; 11.Methanosaeta; 12.Candidatus_Methanoperedens; 13.Methanospirillum; 14.Methanosarcina; 15.Methanobacterium; 16.Methanocella; 17.Candidatus_Nitrososphaera; 18.Candidatus_Nitrocosmicus

图5 最大产甲烷速率和产甲烷延滞期与丁酸互营降解微生物的相关性分析结果

灰色柱子表示Syntrophomonas相对丰度的的相对值, 柱子越高, 其值越大
本文通过共发生网络分析, 探究丁酸互营降解微生物间的共存模式。如图 8 所示, 网络图具有 75个节点和 158 条边, 每个节点代表一个菌属。网络图的平均度为 4.213, 图密度为 0.057, 模块化指数为 0.662。其中, Proteobacteria, Actinobacteria, Ch-loroflexi 和 Firmicutes 为细菌的优势菌门, Euryarchaeota 为古菌的优势菌门。关键分类单元主要包括,,和(图 8(a))。模块 1 主要由丁酸互营氧化菌、产甲烷菌和硝化菌组成, 进一步证明丁酸互营降解过程中互营代谢的重要性(图 8(b))。
3 讨论与结论
3.1 结论
1)丁酸互营降解过程具有以下特征: 产甲烷延滞期随着纬度的升高而增大; 最大产甲烷速率没有表现出明显的地理分布差异; 互营单胞菌()相对丰度较大的样地, 其丁酸完全降解所需时间较短; 互营细菌的相对丰度呈现中国南部明显高于北部的地理分布格局, 环境因子温度对其有关键性影响。
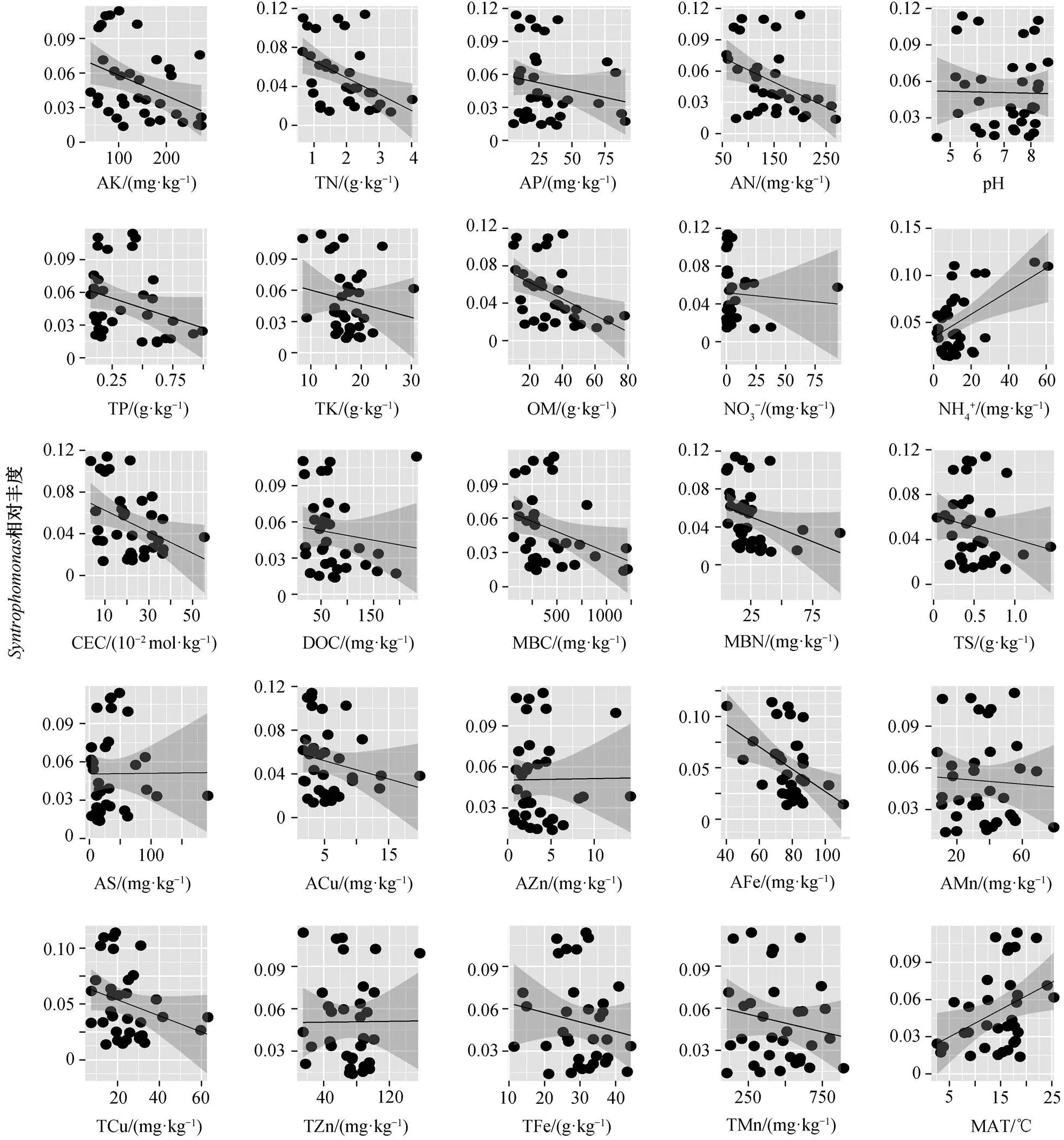
图7 互营单胞菌(Syntrophomonas)相对丰度与环境因子的相关性分析结果
2)丁酸互营降解微生物群落的地理分布格局具有显著的距离衰减关系, 其中细菌群落更加明显, 表明微生物群落的构建同时受到空间距离和环境因子的驱动。
本文分析了丁酸互营降解过程的特征, 揭示了丁酸互营降解微生物的地理分布格局, 研究结果有助于预测不同样地的有机质厌氧分解特性和产甲烷潜力, 为减少 CH4排放提供科学依据。
3.2 讨论
本研究针对丁酸互营降解过程共测定 3 个指标。由于第二代已经历一次富集培养, 产甲烷延滞期大大缩短, 因此采用第一代的产甲烷延滞期进行相关分析。每代的富集培养实验中, 丁酸盐浓度平均测定 4~5 次, 由于测定次数较少, 无法计算丁酸最大降解速率, 因此用丁酸完全降解所需时间来表征丁酸降解过程。
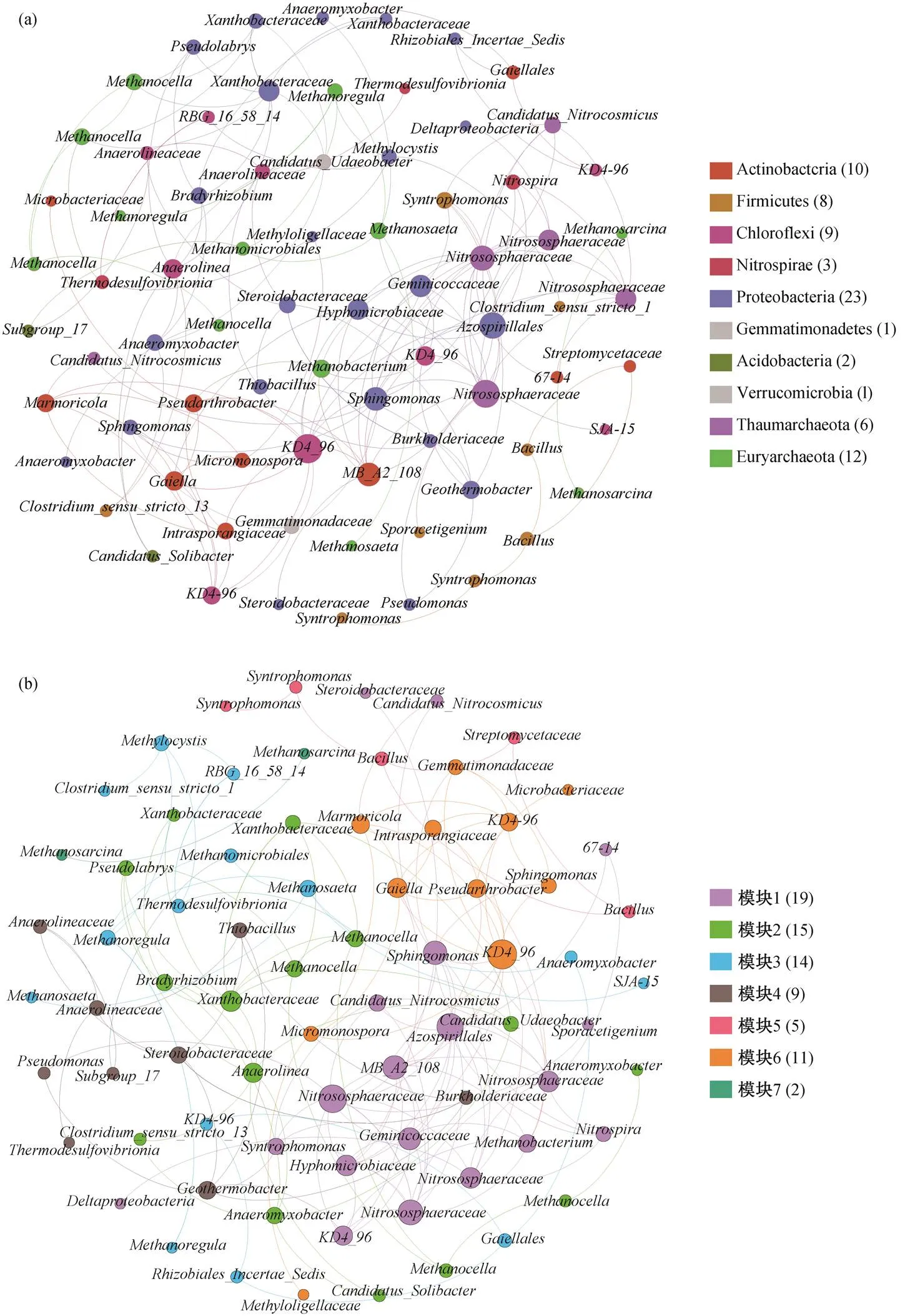
(a)根据微生物门分类对节点着色; (b)根据模块分类对节点着色。图例中括号内数字为节点个数
丁酸完全降解所需时间的分析中排除了两个异常点, 分别是 HD (邯郸, 14 天)和TJ (天津, 13 天)。实验方法部分有关第一代和第二代培养时间的论述中也排除了这两个异常点。两个异常点的产生可能是由于该地点中互营细菌(如)的相对丰度都较高, 而产甲烷古菌的相对丰度相对较低, 相对丰度的差异导致互营效率不一致。
本研究发现的相对丰度随着温度的升高而增大(图 7),相对丰度的地理分布格局也符合这一规律(图 6), 这是由于南部低纬度地区温度较高, 导致相对丰度明显高于北部地区。与此相对应, 中国南部地区产甲烷延滞期低于北部地区, 也是由于南部地区水稻土壤温度相对较高, 土壤中的活性较强, 导致产甲烷延滞期较短。
本研究发现环境因子 pH 和温度对丁酸互营降解微生物群落有重要影响(图 4), 可能由于 pH 是影响土壤有机物降解的关键, 会影响底物利用的有效性[24]。另外, 由于的热力学限制特性, 因此温度对其相对丰度有显著的影响[25]。丁酸降解产氢产乙酸的过程为吸能反应, 受到热力学的限制不能自发地进行[25], 温度升高则有利于缓解热力学限制。这可能是互营单胞菌的相对丰度随纬度升高而降低的主要原因。
鉴于上述的热力学限制特性, 开展实验前, 我们预测温度可能对相对丰度的地理分布格局产生较强的影响。为了验证这种预测, 本文仅对丁酸降解的关键类群展开分析, 对产甲烷的关键类群则没有做详细的论述。在未来的研究中, 可对产甲烷的关键类群进行分析。
丁酸互营降解过程中, 互营细菌与产甲烷古菌之间通常形成团聚体, 以便维持较短的种间距离来保证丁酸的有效降解[26–27]。为了克服该过程中的热力学限制, 产甲烷古菌起到至关重要的作用[2]。本文通过共发生网络分析, 探究水稻土壤中丁酸互营降解微生物的共存模式, 互营细菌和产甲烷古菌被归为同一个模块, 说明微生物类群之间有着密切的互营代谢关系。
本研究采用年平均温度表示样地的温度。实际上, 土壤温度是影响土壤微生物更直接的指标。在未来的工作中, 拟通过直接测定土壤温度来探究温度对微生物群落的影响。
致谢 研究工作得到中国科学院南京土壤研究所褚海燕研究员和冯毛毛同学的帮助, 谨致谢意。
[1] Bouwman A F, Boumans L J M, Batjes N H. Emis-sions of N2O and NO from fertilized fields: Summary of available measurement data. Global Biogeochemi-cal Cycles, 2002, 16(4): 774‒787
[2] Schink B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiology and Molecu-lar Biology Reviews, 1997, 61(2): 262‒280
[3] Schink B, Stams A J M. Syntrophism among prokar-yotes. New York: Springer, 2006
[4] Roderick I. Metabolic activity of fatty acid-oxidizing bacteria and the contribution of acetate, propionate, butyrate, and CO2to Methanogenesis in cattle waste at 40 and 60℃. Applied and Environmental Micro-biology, 1981, 41(6): 1363‒1373
[5] Zou B Z, Takeda K, Tonouchi A, et al. Characteristics of an anaerobic, syntrophic, butyrate-degrading bacte-rium in paddy field soil. Bioscience Biotechnology and Biochemistry, 2003, 67(10): 2059–2067
[6] Wei Z, Jianchao Z, Yahai L. Stimulation of carbon nanomaterials on syntrophic oxidation of butyrate in sediment enrichments and a defined coculture. Scien-tific Reports, 2018, 8(1): 12185
[7] Li H, Chang J, Liu P, et al. Direct interspecies elec-tron transfer accelerates syntrophic oxidation of but-yrate in paddy soil enrichments. Environmental Mic-robiology, 2015, 17(5): 1533–1547
[8] Li F, Tianze S, Wei Z, et al. Stimulatory effect of magnetite nanoparticles on a highly enriched butyrate-oxidizing consortium. Frontiers in microbiology, 2018, 9: 1480
[9] Zhang J, Lu Y. Conductive Fe3O4nanoparticles ac-celerate syntrophic methane production from butyrate oxidation in two different lake sediments. Frontiers in Microbiology, 2016, 7: 1316
[10] Martiny J B H, Bohannan B J M, Brown J H, et al. Microbial biogeography: putting microorganisms on the map. Nature Reviews Microbiology, 2006, 4(2): 102–112
[11] Morlon H, Chuyong G, Condit R, et al. A general framework for the distance-decay of similarity in ecological communities. Ecology Letters, 2008, 11(9): 904–917
[12] Zhao H X, Yang D C, Woese C R, et al. Assignment of fatty acid--oxidizing syntrophic bacteria to syntro-phomonadaceae fam. nov. on the basis of 16S rRNA sequence analyses. International Journal of Systema-tic Bacteriology, 1993, 43(3): 278–286
[13] Jiao S, Xu Y, Zhang J, et al. Environmental filtering drives distinct continental atlases of soil archaea between dryland and wetland agricultural ecosystems. Microbiome, 2019, 7(1): 15
[14] Shi Y, Li Y, Xiang X, et al. Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain. Microbiome, 2018, 6(1): 27
[15] Jin L, Gao X, Du J, et al. Peat bacterial diversity and community structure in Gahai Lake wetlandin Gan’nan. Microbiology China, 2016, 43(11): 2396–2404
[16] Feng Y, Lin X, Yu Y, et al. Elevated ground-level O3negatively influences paddy methanogenic archaeal community. Scientific Reports, 2013, 3: 3139
[17] Edgar R C, Haas B J, Clemente J C, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 2011, 27(16): 2194–2200
[18] Nekola J C, White P S. The distance decay of simila-rity in biogeography and ecology. Journal of Biogeo-graphy, 1999, 26(4): 867–878
[19] Givens D I, Lovegrove J A. Higher PUFA and n-3 PUFA, conjugated linoleic acid,-tocopherol and iron, but lower iodine and selenium concentrations in or-ganic milk: a systematic literature review and meta- and redundancy analyses. British Journal of Nutrition, 2016, 116(1): 1–2
[20] Hiemstra P H, Pebesma E J, Twenhofel C J W, et al. Real-time automatic interpolation of ambient gamma dose rates from the Dutch radioactivity monitoring network. Computers & Geosciences, 2009, 35(8): 1711–1721
[21] Kaikuo X, Changjie T, Rong T, et al. A comparative study of six software packages for complex network research // The Second International Conference on Communication Software and Networks. Singapore, 2010: 350–354
[22] Jiao S, Liu Z, Lin Y, et al. Bacterial communi- ties in oil contaminated soils: biogeography and co-occurrence patterns. Soil Biology & Biochemistry, 2016, 98: 64–73
[23] Barberan A, Bates S T, Casamayor E O, et al. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME Journal, 2014, 6 (4): 343–351
[24] Grybos M, Davranche M, Gruau G, et al. Increasing pH drives organic matter solubilization from wetland soils under reducing conditions. Geoderma, 2009, 154 (1/2): 13–19
[25] Sieber J R, Mcinerney M J, Gunsalus R P. Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annual Review of Microbio-logy, 2012, 66(1): 429–452
[26] Ishii S I, Kosaka T, Hotta Y, et al. Simulating the contribution of coaggregation to interspecies hydrogen fluxes in syntrophic methanogenic consortia. Applied and Environmental Microbiology, 2006, 72(7): 5093–5096
[27] Ishii S, Kosaka T, Hori K, et al. Coaggregation facili-tates interspecies hydrogen transfer between-and-. Applied and Environ-mental Microbiology, 2005, 71(12): 7838–7845
Biogeographic Patterns of Microbial Communities Associated with Syntrophic Butyrate Degradation in Paddy Soils in Eastern China
FEI Yuanyuan, JIAO Shuo, LU Yahai†
College of Urban and Environmental Sciences, Peking University, Beijing 100871; † Corresponding author, E-mail: luyh@pku.edu.cn
The authors collected 34 paddy soil samples along the latitude from eastern China. Enrichment experiment was conducted under anaerobic conditions with sodium butyrate as the sole substrate. The authors investigated the microbial community characteristics and the functional activity of syntrophic butyrate degradation and the biogeographic patterns of relative abundance ofin these soils by Illumina sequencing of 16S rRNA genes. The lag phase of CH4production (3–14 days) increased towards higher latitudes, whereas the maximum rate of CH4production did not. The correlation analysis on influencing factors revealed that-was the key syntrophic bacterial taxon associated with butyrate degradation and its relative abundance was significantly influenced by mean annual temperature (MAT). The sampling sites with a relatively high abundance ofhad a shorter time for the complete degradation of butyrate. Distance-decay patterns cha-racterized by a steeper slope were found in microbial communities associated with syntrophic butyrate degradation (<0.05), indicating that the construction of microbial communities was driven by both spatial distance and envi-ronmental factors.
butyrate degradation; methanogenic production; relative abundance of; microbial biogeography
10.13209/j.0479-8023.2020.109
2019‒12‒23;
2020‒04‒25
国家重点研发计划(2016YFD0200306)和国家自然科学基金(41630857)资助
