2,5-二甲基四氢呋喃的燃烧反应动力学机理研究
2021-02-01李德华陈日新程晨王锡斌
李德华,陈日新,程晨,王锡斌
(西安交通大学能源与动力工程学院,710049,西安)
四氢呋喃族燃料属于第二代生物质燃料,可以利用秸秆、玉米芯、木屑等农林业废料为原材料制取,相比于乙醇等第一代生物质燃料,四氢呋喃族和二氢呋喃族燃料在生产过程中不消耗农业资源。如果作为清洁可替代的燃料,可充分利用农林废弃物大规模生产,对于降低环境污染、减少国内石油的供应压力、降低发动机排放物及增加农业产品附加值和农民收入都具有重要意义。四氢呋喃族燃料辛烷值比汽油低,在新型燃烧模式例如HCCI中具有很大的应用潜力。同时,研究发现该类组分是众多烃类燃料燃烧的中间产物[1-6],对烃类燃烧反应具有相当的影响。目前,有关四氢呋喃燃烧机理的研究偏少,有必要开展四氢呋喃族燃料燃烧的反应动力学机理的研究。
目前,关注较多的该族燃料主要有四氢呋喃(THF),2-甲基四氢呋喃(MTHF),2,5-二甲基四氢呋喃(DMTHF),其中四氢呋喃和2-甲基四氢呋喃的高温反应模型都已经比较成熟。Tran等搭建了THF的高温燃烧模型[7];Moshammer等开发了第一个MTHF的高温燃烧模型[8];Tripathi等开发了MTHF低温燃烧模型[9];Bruycker等开发了MTHF的裂解和燃烧模型[10],在关键速率系数上更加准确;Fenard等开发了MTHF的低温燃烧模型[11],通过速率规则和类比的方法给出重要速率系数,略微粗糙,而后又对该低温燃烧模型的上述问题加以完善[12]。到目前为止,还没有公开发表的DMTHF反应模型,相对于THF与MTHF,DMTHF的优势在于分子结构复杂,其反应模型可通用于THF和MTHF,所以有必要建立DMTHF的反应模型。
本文在获得DMTHF燃烧反应相关的热力学、化学动力学和输运数据库之后,基于Bruycker有关MTHF的反应模型[13]对DMTHF的燃烧过程进行了反应动力学建模,得到了一个对THF、MTHF和DMTHF通用的高温氧化机理,之后借助该模型对DMTHF燃烧的反应路径和反应敏感性进行研究。
1 DMTHF反应模型的确定
1.1 热力学参数的确定
一个反应系统的模型主要包含3个数据库,热力学、化学动力学和输运数据库。底层的C0~C4的反应及其相关组分的特性研究已经很深入,所以需要确定的主要是DMTHF起始反应涉及的组分,即DMTHF及其一级反应产物,包括脱氢反应、开链反应及异构化反应产物的热力学参数。图1给出了脱氢位置示意图,随脱氢位置的不同,各脱氢产物分别定义为DMTHF-MJ、DMTHF-2J、DMTHF-3J。

图1 脱氢位置示意图
开环反应的4种中间产物的生成路径如图2所示,4种反应产物分别用P1、P2、P3、P4表示。

图2 DMTHF的二级产物的生成路径
DMTHF燃烧过程中会不可避免地产生部分二氢呋喃类物质,2,5-二甲基-2,3-二氢呋喃(DMDHF)就是其中生成较多的物质之一,同时会发生较为典型的异构反应,如图3所示。

图3 DMDHF的异构反应
使用Gaussian09程序包[14],采用CBS-QB3方法对以上各物质组分的分子结构、分子热力学数据和分子能量数据进行计算,然后利用THERM[15]软件基于基团贡献法计算气相分子热力学数据,根据分子结构、生成焓、振动频率、转动惯量、分子对称性以及校正因子等计算分子的生成焓、熵以及比热容等热力学数据。表1为计算所得部分热力学参数。

表1 DMTHF分子自由基的生成焓和键能
1.2 反应化学动力学参数的确定
DMTHF燃烧模型的建立中最为重要的就是确定燃料分子的氧化路径,并且给出每一步基元反应的速率系数。本文中大多数速率系数的来源都是参考Tripathi和Bruycker的MTHF的模型,不过对于少数重要反应,需要更为准确的速率系数,可通过理论计算得到,主要包括氢提取反应、裂解反应和异构反应。反应速率常数的计算理论主要采用了过渡态理论(TST)和RRKM理论,考虑势能面和反应物浓度的影响。TST理论认为反应物要转变为过渡态后生成产物,势能面的差异决定了反应速率。RRKM理论认为反应物在发生化学反应之前,肯定会先一步经过碰撞活化或去活化,而碰撞情况与反应物分子的浓度有关,即表观反应速率会与压力相关。
1.2.1 氢提取反应 如前所述,DMTHF可发生3种氢提取反应R1、R2、R3,经过量子化学计算得到DMTHF-MJ/2J/3J以及相关的几种中间产物TS1、TS2、TS3的反应势能面,如图4所示。氢提取的难易与相应位置处的C—H键能密切相关,DMTHF分子M位的C—H键能最高,3号次之,2号最低。

图4 DMTHF氢提取反应的势能面
图5展示了DMTHF与H发生的3种氢提取反应的速率和分支比(表示不同反应路径几率的比例关系)。2号位的氢提取占据了主导地位,随着温度的升高,能垒的影响会减弱,2号位氢提取的主导地位也逐渐下降,在2 500 K其分支比降低到了50%。这主要是由于M位和3号位的氢提取反应能垒较高,活化能较大,导致速率对温度更加敏感。任何情况下,2号位的氢提取产物DMTHF-2J的生成量都会更多。M位和3号位的氢提取速率相近,其分支比也非常接近。

(a)反应速率(b)分支比图5 DMTHF与H发生的3种氢提取反应的速率和分支比
1.2.2 裂解反应 DMTHF分子的裂解开环反应包含了DMTHF分子的裂解反应和DMTHF中间基的裂解开环反应。裂解反应能垒较高,只有在缺氧或高温下才能产生较大影响。DMTHF分子的主要裂解反应如图6所示,图7给出了DMTHF的几种裂解反应的速率随温度和压力的变化。
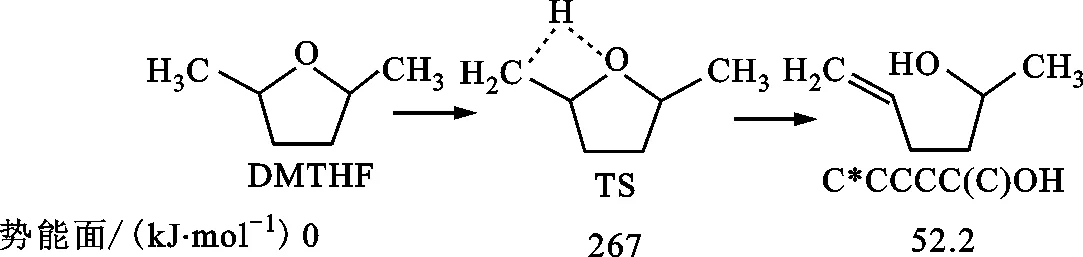
图6 DMTHF的燃料分子裂解反应
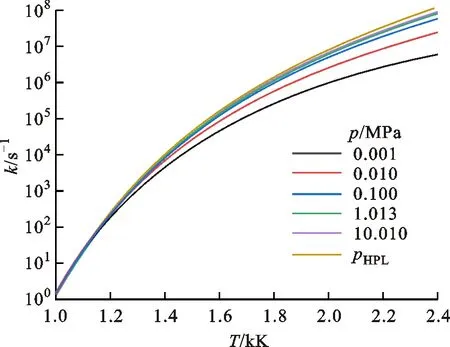
图7 DMTHF裂解反应速率随温度和压力的变化
作为单分子反应,其反应速率对压力敏感,随着压力的增加,反应速率会升高,但升高速率越来越慢,最终达到接近高压限值pHPL。DMTHF在经过氢提取反应形成自由基之后,会经历β裂解形成开环自由基,而后开环产物会进一步β裂解形成C4及以下的小分子物质。Simmie着重研究了DMTHF-2J和DMTHF-3J的相关势能面[16],对DMTHF-MJ的反应路径计算较少,故本文在此将主要工作放到了DMTHF-MJ的氧化路径上。图8给出了DMTHF-MJ相关反应的势能面和反应路径R4、R5,其中反应路径R5的能垒很高,故反应速率比较小,如图9所示。因为R5的反应速率较小,所以其分支比也较小。
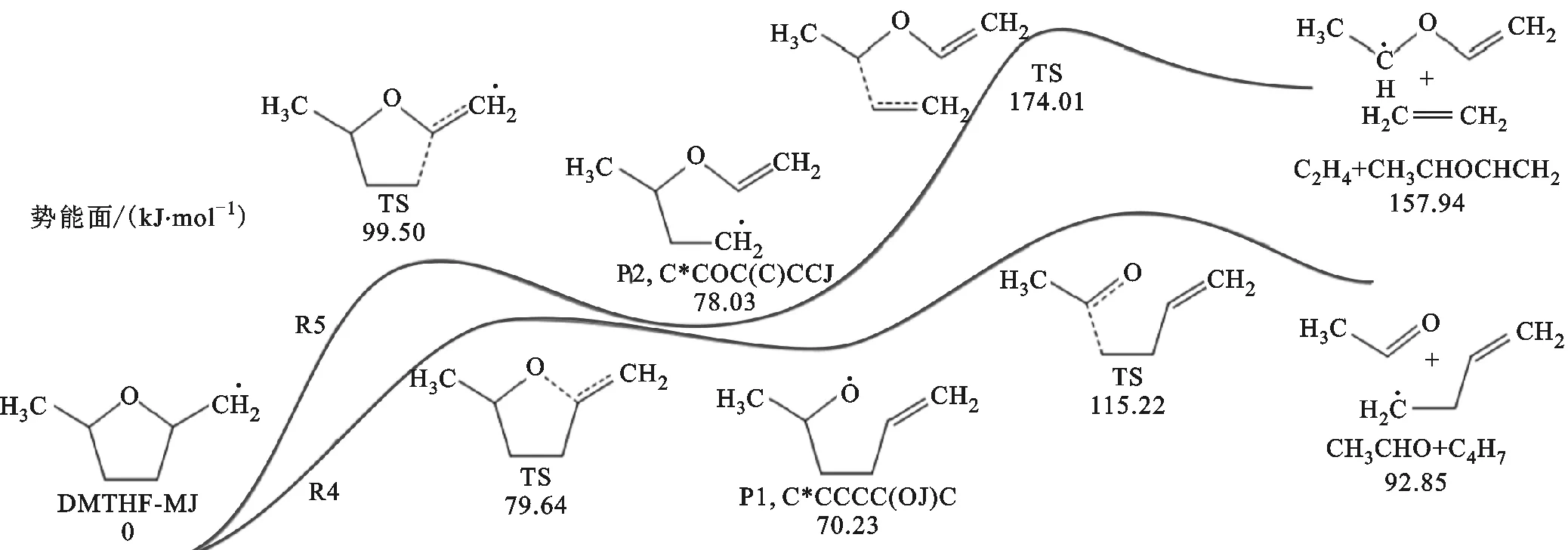
TS—过渡态。图8 DMTHF-MJ的势能面
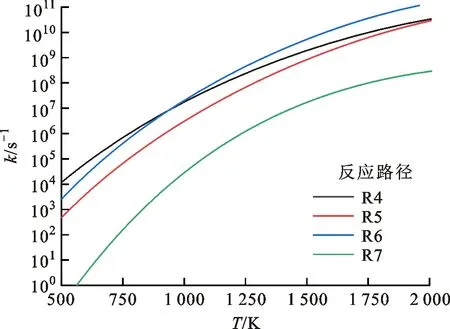
图9 DMTHF-MJ裂解反应速率随温度的变化
2 模型验证
通过上面研究得到DMTHF氧化的反应动力学机理,本文利用CHEMKIN Pro软件中的PREMIX模块,根据上述反应机理和热力学数据、输运数据等计算层流火焰速率以及对低压预混火焰中组分浓度分布进行模拟计算。
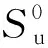
(a)0.1 MPa下的结果

(b)423 K下的结果图10 DMTHF层流燃烧速度的实验值和模拟值对比
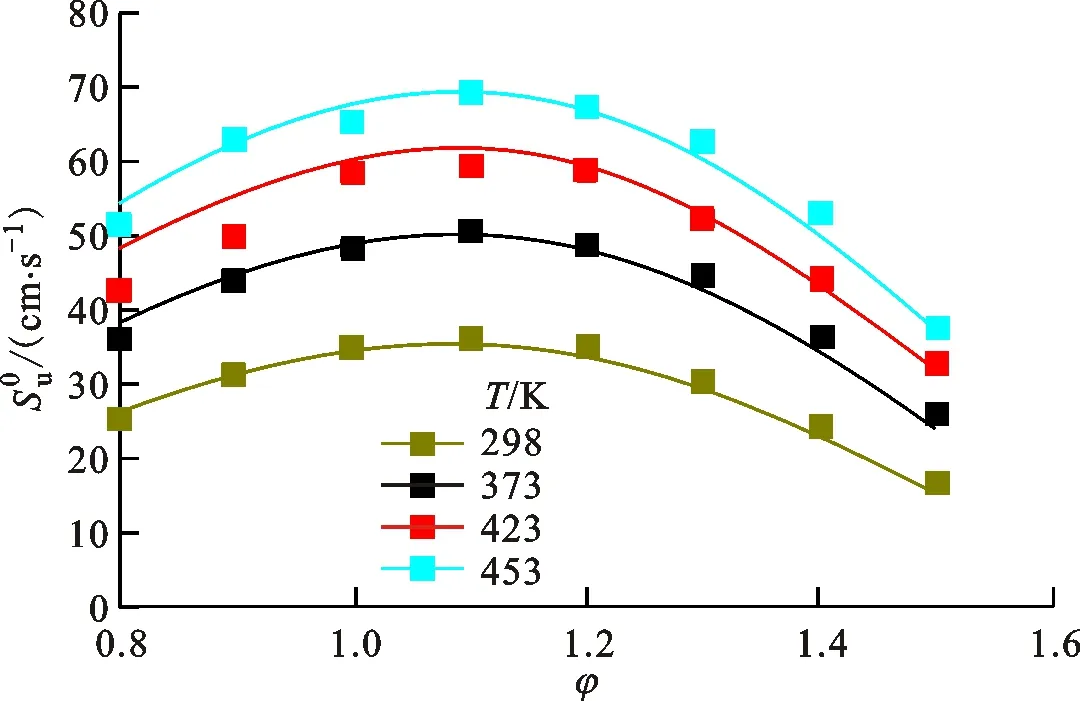
3 DMTHF的路径分析
图11展示了初始温度为453 K、初始压力为0.101 MPa、当量比分别为0.8和1.3时DMTHF的反应路径分析。与MTHF和THF有较大不同的是,DMTHF的层流燃烧中存在一定量的裂解开环反应,在大当量比时分支比接近了10%,而这类反应在THF和MTHF中的存在量都非常不明显。其主要原因是DMTHF甲基上的H与O之间距离较短,容易形成周环反应,从而形成较大的反应速率。由于分子对称性的原因,DMTHF的分子自由基数量和路径较MTHF减少,不过对于M位和C2位的自由基,其反应机制与THF和MTHF基本一致,都是快速通过β裂解开环,并发生第二次β裂解生成C2和C3小分子。根据键能的大小,可以预测C2和C3反应路径的分支比。DMTHF-3J的反应路径也较为多样,通过消氢与消甲基等反应形成了较大比例的二氢呋喃族物质,这个现象在MTHF和THF中也有发生。这类二氢呋喃物质经过了比其他中间产物更复杂的过程(氢提取、异构、裂解)消耗掉。此处需要说明,由于对二氢呋喃类(DHFs)物质并没有较多的实验研究,因此相关基元反应的速率或许存在较大的不确定性,导致DHF反应系统对整体反应机理的准确性有一定的影响。随着当量比的变化,燃料除裂解反应以外的消耗途径的分支比变化并不明显。
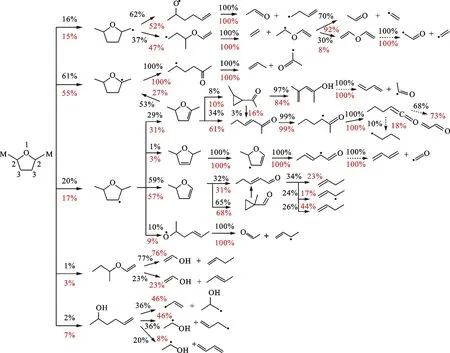
百分数—分支比;黑色—φ=0.8;红色—φ=1.3。图11 DMTHF的路径分析
DMTHF分子环上的甲基更多,反应类型也更为多样,其复杂性主要表现在燃料分子裂解反应比例的增多、一级和二级中间产物的增多。除了普通裂解反应以外,DMTHF-3J可以通过β裂解脱甲基,也可以在2号位消氢,消耗路径较多。因此,在火焰中,THF-3J和MTHF-3J/4J的浓度峰值都较高,而DMTHF-3J浓度峰值较低。这说明脱氢生成二氢呋喃类物质是受到了甲基较大的影响,究其原因在于甲基的存在使得C2—H键能降低,同时甲基与环之间的C—C键能也比较低,容易发生β裂解。
图12展示了初始温度为453 K、初始压力为0.1 MPa时DMTHF火焰的敏感性分析。DMTHF的重要反应依然是以小分子物质为主。C2类物质更为丰富,THF和MTHF中重要的C2类小分子在DMTHF中都有表现,主要有不饱和烯烃及自由基C2H2、C2H3、C2H4、C2H5,醛类及自由基CH2CHO、CH2CO、CH3CO、HCCO。可以看到,图中有3个包含C3H6和AC3H5的反应,其中一个是AC3H5加氢生成C3H6的反应(自由基加成反应能垒约为0,反应发生非常容易),另外两个是C3H6氢提取生成AC3H5的反应
C3H6+OH=AC3H5+H2O
C3H6+H=AC3H5+H2
AC3H5+H=C3H6
这3个反应组成了一个不断消耗H和OH的系统,因此在不同的当量比下都是负的敏感性,而竞争反应C3H6+H=C2H4+CH3虽然消耗了H并生成了CH3,但是依然有正的敏感性。由于生成的DMTHF-2J几乎全部发生裂解反应生成C3H6,是火焰中丙烯的主要来源,因此该反应有非常大的负敏感性。
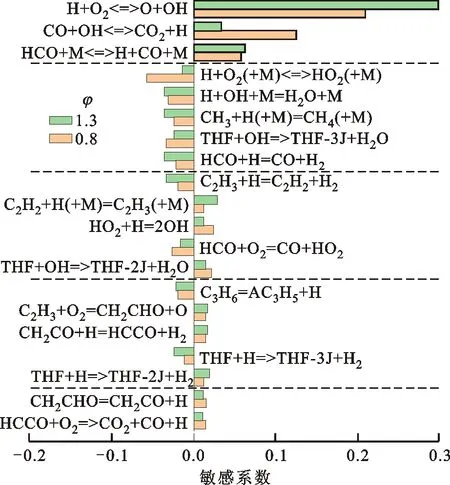
图12 DMTHF火焰的敏感性分析
4 结 论
(1)本文结合高精度电子结构计算理论和基元反应统计速率理论,对DMTHF的高温燃烧模型进行了构建,最终得到了一个可以用于THF、MTHF和DMTHF的反应动力学模型,通过与不同温度、压力、当量比下的DMTHF层流实验研究结果进行对比,得到了基本吻合的结果,模型获得了验证。
(2)DMTHF的3种氢提取反应中,2号位的氢提取占据了主导地位,任何情况下2号位的氢提取反应产物DMTHF-2J生成量都更多,M位和3号位的氢提取反应速率相近,分支比也十分接近。对裂解反应的研究表明,DMTHF-MJ的氧化路径中,生成P1的反应占据了主导地位。
(3)DMTHF分子环上有更多的甲基,因此反应类型也更为多样,使得敏感性较高的反应中,涉及到的重要中间产物也非常丰富。反应路径的复杂性变化主要表现在燃料分子裂解反应比例的增多、一级和二级中间产物的增多。重要中间产物更加丰富主要表现在C2类分子上。
(4)DMTHF燃烧反应中,DMTHF-2J所占的分支比更大,且后续路径中会生成大量的C3H6,而与C3H6相关的反应也存在很显著的负敏感性,这说明C3H6及自由基AC3H5之间不断消耗H和OH的反应系统对DMTHF的反应强度产生了很大的负面影响。
