架起化学-酶催化之间的桥梁:构建策略及催化应用
2021-01-29栾鹏仟周丹丹王晓天陈冉高士耆赵浩黄琛刘运亭高静姜艳军
栾鹏仟,周丹丹,王晓天,陈冉,高士耆,赵浩,黄琛,刘运亭,高静,姜艳军,2
(1 河北工业大学化工学院,天津300130; 2 河北工业大学化工节能过程集成与资源利用国家地方联合工程实验室,天津300130)
引 言
化学催化在催化领域一直占据着主导地位,尤其在精细化学品[1-3]、先进材料[4-6]、电池[7-9]等领域得到广泛应用,为人类社会进步做出极大的贡献。然而,化学催化仍然存在一些不可避免的缺点,如环境污染、选择性差、反应条件苛刻等。近年来,生物催化以其反应条件温和、环境友好以及(化学、区域和立体)选择性强等优点成为研究热点,在一定程度上能够弥补化学催化的不足[10-11]。尽管如此,生物催化同样也存在着许多局限,其中最为突出的是生物催化剂的稳定性、催化反应的多样性、重复使用性等都远不如化学催化剂,并且制备条件复杂,周期长。将化学催化与生物催化相结合,实现两者的优势互补,并用于设计全新、精巧的反应来合成高附加值产品,逐渐成为新的研究领域[12-13]。自1997 年Bäckvall 等[14]首次将水解酶和金属催化剂结合在一起,化学-酶级联催化至今已经取得了极大的进步。
传统的化学-酶催化转化通常是逐步进行的,即完成第一步反应后需要通过复杂的分离纯化技术将中间产物从体系中分离,然后才能进行接下来的反应。这种方式的化学-酶催化不仅费时费力,而且增加了整个工艺的成本。为解决这些弊端,研究者开发了一锅法化学-酶级联催化反应,所有反应均在同一个容器中进行,省略烦琐的分离纯化步骤,从而简化工艺降低成本[15-17]。
尽管工业上使用化学-酶催化转化的实例正在不断增长,但其内在的复杂性严重阻碍了这一概念的广泛应用,其中最大的障碍是化学催化与生物催化的不相容性。这种不相容性包括不同催化剂自身的不相容和催化条件差异导致的不相容。催化剂自身的不相容性主要表现在一锅法转化中化学催化剂与酶的直接接触导致催化剂相互失活。例如,Latham 等[18]在结合卤化酶和金属钯(Pd)的研究中发现,酶的氧化还原性和亲和力会严重影响Pd的活性。而反应条件的差异性表现得更为明显,酶催化一般需要在条件较为温和的水相介质中发生,而大多数化学催化剂不仅要在高温强碱下发挥作用,而且还需有机溶剂作为反应介质。在这种苛刻的环境下,大多数的酶都很难维持原有的催化活性,例如酚酸脱羧酶(PAD)在疏水溶剂中是有活性的,但是长时间暴露在甲苯溶剂中会导致其完全失活[19],而甲苯正是大多数化学催化剂较为理想的反应介质。化学催化剂和生物催化剂任何一方的失活都将直接导致化学-酶催化转化的失败,因此需要发展有效的策略来应对不同原因导致的不相容。
基于以上背景,本文总结了克服化学催化与酶催化不相容问题的方法和策略,包括时间分隔和空间分隔策略。另外,还介绍了化学-酶级联催化在手性化合物动态动力学拆分和药物分子(或中间体)合成等方面的应用。最后,对未来化学-酶级联催化的发展方向进行展望。
1 化学-酶级联催化构建策略
催化条件(如温度和pH)引起的不相容,决定了两种催化反应不能同时发生。因此一般采取时间分隔的方式,第一步反应结束后,调节温度和pH 到下一步反应所需的范围后,再加入催化剂与试剂进行后续反应。对于溶剂带来的问题可采用两相体系和非常规溶剂的方法来解决。催化剂不相容的问题通常采用空间分隔的策略来解决,具体方法包括将不同催化剂各自固定化、制备分隔式集成催化剂或借助高分子膜来分隔催化剂。通过这些手段均可有效实现不同催化剂的活性中心在空间上的分离,从而避免接触失活。
1.1 催化条件不相容
1.1.1 时间分隔(temporal separation) 一锅化学-酶级联反应分为三类,分别为连续、并行和协同反应。在反应过程中需要调节体系温度、pH、底物浓度等条件的反应为典型的连续反应。与其他两类反应相比,连续反应过程需要进行额外的操作,譬如添加反应组分或调节反应条件等,使得过程更加烦琐,但是该方法可以将更多催化条件相差很大的催化剂级联在一起,创造出更多全新的一锅化学-酶催化反应,因此应用最为广泛。
González-Sabín 等[20]通过连续反应的方式实现了钌(Ru)基催化剂和转氨酶(ω-TA)的结合。如图1(a)所示,Ru催化的烯丙醇异构化在50℃条件下进行,而ω-TA 却不能耐受50℃的高温条件。此外,在研究中发现底物烯丙醇对转氨酶的活性存在一定程度的影响。因此,在化学催化完成之后,先降低温度到转氨酶耐受的范围,将体系稀释使底物浓度降低从而减小对酶活的影响,最后再将转氨酶引入体系,进行后续的生物催化。由此可看出采取连续反应的方式进行一锅化学-酶催化转化可以有效地消除催化条件差异带来的不相容问题。
金属催化的C—C 交叉偶联反应已被公认为有机化学中形成C—C 键最有力和最通用的合成工具之一,如Suzuki、Heck 和Negishi 反应。这类反应最吸引人的一个优点在于其可在水相中进行,有利于与生物催化的结合。Groger 等[21]首次将Pd 催化的Suzuki交叉偶联反应与醇脱氢酶(ADH)催化的酮还原反应相结合,并成功地在水相中完成了二者的级联反应,如图1(b)所示。在70℃的碱性水溶液中,Pd(PPh3)Cl2催化苯硼酸与对溴苯乙酮发生C—C 交叉偶联反应。反应结束后无须分离中间产物,待体系温度降为室温、调节pH 到中性后加入ADH 进行后续的还原反应,最终生成一系列手性二芳醇。Garg等[22]用类似的方法在水中级联制备抗阻胺药物奥非那汀的对映体。Turner 等[23]开发了一种一锅化学-酶级联反应,采用了时间分隔的连续反应方式,最终得到一系列光学纯的N-保护的非天然L-和D-二芳基丙氨酸衍生物。
除了前面介绍的交叉偶联反应外,González-Sabín 等[24]采用连续反应的方式成功地在水相中将KRED 催化的不对称还原反应与金属催化的炔烃环异构化反应相结合,以较高的产率合成了1,4-二醇、内酯、γ-羟基羰基化合物(羧酸、酯和酰胺)等多种对映体纯化合物,如图2(a)所示。同年该课题组又建立了外消旋醇不对称转化为胺的化学-酶级联催化体系[25],如图2(b)所示。将有机催化剂2-氮杂金刚烷-N-氧自由基(AZADO)、氧化剂次氯酸钠(NaOCl)和转氨酶结合,在水介质中实现了一锅氧化-胺化连续过程。该方法具有宽广的底物谱,包括传统的仲醇和空间位阻较大的β-取代环烷醇。其中高度立体选择性的不对称生物胺化使整个反应过程具有非常高的产率、光学纯度和非对映选择性(产率>90%,ee>99%,dr>49∶1)。
化学-酶级联反应中催化剂的不相容性及其交叉反应性问题频繁出现,使得大多数例子都是连续的一锅反应,而不是真正的并行反应。但是连续反应也存在一些局限性,比如反应过程中涉及可逆反应或极不稳定的中间体时,该方法则不适用。因此,需要发展其他的策略,以解决这些问题或实现更为高效的并行反应。
1.1.2 两相体系 除了温度和pH 引起的催化条件不相容外,溶剂引起的不相容性同样不可忽视。绝大多数化学催化剂偏好有机介质,在水相中很难正常发挥催化活性,但几乎所有的酶对有机溶剂是不耐受的。而且,有机试剂在水相中的溶解度普遍较差,这大大增加了使用水作为反应介质的难度。因此,研究者利用两相体系解决溶剂带来的不相容问题以及底物溶解性问题[26-27]。该体系由两种互不相溶的液体组成,化学催化剂溶解在有机相中,而酶溶解在水相中,从而有效地缓解溶剂带来的不相容问题。从另一个角度上考虑,两相体系在一定程度上也能限制不同催化剂之间的接触,避免催化剂发生接触失活。

图2 水溶液中化学-酶催化不对称级联合成1,4-二醇、内酯、γ-羟基羰基化合物[24(]a)及2-(苄氧基)环烷胺[25(]b)Fig.2 Chemoenzymatic for the asymmetric synthesis of 1,4-diols,lactones,γ-hydroxy-carbonyl compounds[24](a)and(benzyloxy)cycloalkanamines[25](b)in aqueous medium
Castagnolo 等[28]开发了一种由水和异辛烷组成的两相体系用于一锅化学-酶催化合成吡咯,如图3(a)所示。首先底物和Ru 基催化剂溶于异辛烷中,酶溶于缓冲溶液中。随着化学催化的不断进行,生成的中间产物3-吡咯啉不断地向缓冲溶液中扩散,随即在单胺氧化酶(MAO-N 或6-HDNO)的作用下发生芳构化反应,从而在水相中生成吡咯。而且,生成的吡咯又不断地从水相扩散到有机相中,促进了反应平衡向产物方向的移动,这是该反应在温和条件下高产率地合成吡咯的原因。实验结果表明该两相体系解决了溶剂引发的不相容问题,而且还避免了化学催化剂与酶发生络合作用。最近,该课题组又应用该两相体系,以脂肪族二烯丙基醚为原料,结合Ru基催化剂催化的烯烃复分解反应和漆酶(laccase)催化的芳构化反应,通过一锅法化学-酶级联催化合成了一系列呋喃类化合物[29],如图3(b)所示。由异辛烷与缓冲溶液组成的稳定的两相体系还被应用于其他一锅化学-酶级联反应中[30-31]。

图3 两相体系一锅化学-酶催化合成吡咯[28](a)和呋喃[29](b)类化合物Fig.3 One-pot chemoenzymatic synthesis pyrroles[28](a)and furans[29](b)compounds though biphasic system
蔗糖磷酸化酶(SP)能够催化蔗糖可逆地磷酸化为α-D-葡萄糖-1-磷酸和D-果糖,其广泛的受体特异性被开发用于葡萄糖向多种受体分子转化,如多元醇、酚类、羟基呋喃酮和二苯乙烯类化合物。然而由于SP 对这些底物的低亲和力(Km>1 mol/L),通常情况下需要向体系中加入二甲亚砜或甲醇等助溶剂来提高底物的溶解度。助溶剂的浓度对反应影响很大,浓度过低时不足以溶解足够的底物,而浓度过高时会直接抑制酶的活性甚至使其失活。Winter 等[32]利用由缓冲溶液和乙酸乙酯组成的两相体系通过化学-酶法合成α-D-葡萄糖苷。水相中含有酶和水溶性底物,而疏水性底物则溶解在有机相中。通过搅拌或摇动使这些底物由有机相转移到水相,并在水相中发生酶催化转化。通过对比产物2-O-α-D-葡萄糖苷邻苯三酚在助溶剂体系和两相体系中的转化率,发现后者的转化率提高了5 倍之多,有效地抑制了副产物的产生。
手性亚砜是一类具有生物活性的重要化合物,其中一些亚砜是市售药物的活性成分,如奥美拉唑[33]。此外,它们还可作为不对称合成中的手性配体、催化剂和构建模块[34]。Míšek 等[35]在由缓冲溶液和正癸烷组成的两相体系中首次开发了对手性亚砜进行去消旋化反应。在该反应体系中,酶可以耐受不同的取代基,底物的适用范围较广,为合成对映体纯亚砜提供了一条可行的途径。
PAD 是典型的在有机溶剂中失活的酶,而且催化的底物在水中溶解度极低。Kara等[36]将PAD的脱羧反应与Pd催化的对羟基苯乙烯氢化反应耦合,在由缓冲溶液和正己烷组成的两相体系中合成4-乙基愈创木酚。然而这种传统的两相催化体系存在一个很明显的缺陷,反应仅发生在表面积有限的液面之间,导致反应速率缓慢。解决这类问题最直接的方法是增大液面之间的接触面积。Kourist等[37]将PAD 封装于聚乙烯醇(PVA)/聚乙二醇(PEG)冷冻凝胶中与化学催化剂催化的烯烃复分解反应级联制备具有强抗氧化活性的4,4′-二羟基二苯乙烯,如图4(a)所示。封装后的PAD 不仅可在叔丁基甲醚(MTBE)中正常进行催化,而且凝胶还增大了液面的接触面积。底物从有机相进入水相,催化得到的中间产物重新进入有机相。分离PAD 凝胶后,添加化学催化剂进行后续的烯烃复分解反应。
如上所述,将酶封装于“油包水”的结构中可以实现有机相中的酶催化反应。与之类似,将化学催化剂封装于“水包油”的结构中可以实现水相中的化学催化反应。TPGS-750-M 表面活性剂在水中可形成内部疏水外部亲水直径约50 nm 的球形胶束,内部的疏水核由维生素E 组成,可装载过渡金属催化剂和亲脂底物,如图4(b)所示。反应发生在这些特制的纳米反应器内部的疏水核心,并且生成的产品在胶束之间可通过水进行动态交换,因此存在与酶构建级联反应的潜力。Lipshutz 等[38]利用TPGS-750-M 形成的胶束实现了金属催化反应(Pd 催化Heck偶联反应、Au/Ag催化炔烃水合反应和Rh催化1,4-加成反应)与生物催化反应(ADH 催化还原反应)的结合,合成了一系列光学纯的手性仲醇。其中,金属催化的有机反应发生在胶束内部,而酶催化发生在与胶束相容的水介质中。这类设计可有效地将金属催化剂限制在有机相中,从而避免外部水相对金属的干扰,同时也阻断了酶与有机相的接触,使酶能够一直在水相中维持较高的活性。水中存在的纳米胶束不仅可为化学和生物催化提供反应介质,而且还可作为底物、产物和催化剂的贮存器,起到降低非竞争性酶抑制的效果。
1.1.3 非常规溶剂(non-conventional media) 两相体系在一定程度上可以缓解溶剂带来的不相容问题,然而低产率和内在的复杂性严重限制了两相体系在间歇反应中的应用。近年来,非常规介质的发展给上述问题带来了新的解决方案[39]。非常规介质包括:无溶剂过程、超临界流体(SCFs)、生物质衍生溶剂、氟化溶剂、离子液体(ILs)和低共融体(DESs)。本节将重点介绍非常规溶剂用于解决一锅化学-酶催化过程中溶剂引起不相容问题的研究进展。
ILs 是由有机阳离子和有机或无机阴离子组成的低熔点盐,研究表明ILs 能够通过产生的微环境稳定不同种类的酶,并起到液体载体的作用。此外,使用ILs 作为反应介质,在一定程度上能够增强生物催化剂的对映选择性和催化剂活性,并使其具有更高的操作稳定性。Schmitzer 等[40]在由含ILs 和水相组成的两相体系中,实现了Pd 催化的Suzuki偶联与ADH 催化的生物还原反应的级联。Suzuki 偶联发生在ILs 相中,ILs 可增强金属催化剂的催化活性和稳定性。生成的中间产物转移到水/IL 界面上被ADH 还原,最终以高收率和高对映选择性合成了一系列手性二芳醇。

图4 (a)PAD封装于PVA/PEG凝胶中用于一锅化学-酶催化反应[37];(b)TPGS-750-M在水相中形成的胶束[38]Fig.4 Encapsulate PAD in PVA/PEG for one-pot chemoenzymatic cascade reaction[37](a);The designer surfactant TPGS-750-M forms micelles in water[38](b)
DESs 是由廉价的生物可降解氢供体和氢受体组成的混合物,作为一种新型绿色溶剂在生物催化、金属催化、有机合成和材料化学等领域得到了广泛的应用。González-Sabín 等[41]首次将DESs 应用于酮的不对称胺化中。ω-TA 在75%(质量)DESs和缓冲液组成的反应介质中具有很好的稳定性。将Pd 催化的Suzuki 交叉偶联反应与异源外生菌ATA(EX-ωTA)催化的胺化反应级联,实现了在该混合介质中合成双芳胺。DESs 的增溶特性使金属催化过程中底物的浓度和酶催化过程中中间产物的浓度分别升高至200 mmol/L 和25 mmol/L。与之前的研究相比,DESs的引入提高了产品12%的收率。并且ω-TA 在该混合溶剂中所展现出的超高酶活,无疑突出了生物可再生溶剂的实际应用价值。Wang等[42]通过在两相体系中加入DESs,得到了稳定的微乳液。由于DESs降低了溶液的表面张力,加之脂肪酶的界面活化表现出的高活性导致产物的合成速率提高了六倍之多。
Capriati 等[43]将Ru 催化的烯丙醇异构化与KRED 的不对称生物还原进行耦合,制备出一系列手性醇化合物。KRED 在由DESs 和缓冲溶液组成的混合溶液中表现出很好的稳定性与催化活性,而且研究发现混合介质中DESs 的含量越高,KRED 的对映选择性越强,从而所得仲醇的光学纯度越高。同年该课题组又以DESs 和缓冲溶液为反应介质获得一组产率高、对映体纯(ee>99%)的手性双芳醇[44]。
1.2 催化剂自身不相容
除了不同催化条件引起的不相容,催化剂自身不相容也是建立化学-酶催化体系时常见的障碍。解决此类问题最常用的方法是将催化剂空间分隔(space separation),阻断活性中心在空间上的接触。空间分隔的方法分为:独立固定化催化剂、分隔式集成催化剂和膜分隔。本节将对近年来研究者通过制备各种先进材料应用于化学-酶催化反应中解决催化剂自身不相容问题的研究进行详细介绍。
1.2.1 独立固定化催化剂 将化学-酶催化反应中的某一种催化剂采用固定化技术固定于载体中,将其限制在载体内部。同时借助载体上的孔阻止另一种催化剂进入载体内部,使二者在空间上无法接触。载体的选择对催化剂的性能有着至关重要的影响。一般来说,载体的选取需要遵循以下几个原则:(1)比表面积大,有足够的空间容纳催化剂;(2)稳定性高,不仅要具有很高的机械稳定性以免材料塌陷,还要具有很好的化学稳定性避免在反应过程中引发副反应;(3)制备过程简单;(4)环保无毒;(5)成本低廉。
介孔硅基材料[45-46]凭借着高稳定性、低毒性和高比表面积等优势在众多材料中脱颖而出。Inagaki等[47]在研究中发现Rh 配合物与牛血清白蛋白(BSA)的直接接触会严重影响Rh 的活性。于是他们将Rh 配合物固定于联吡啶基周期性介孔有机二氧化硅(BPy-PMO)中,介孔的存在使蛋白质无法进入BPy-PMO 内部与Rh 接触。随后将Rh@PMO 与马肝醇脱氢酶(HLADH)结合,用于合成(S)-4-苯基-2-丁酮。由于避免了催化剂的失活,产品具有较高的转化率和对映选择性,并且Rh@PMO在2-环己烯-1-酮转移加氢反应中表现出较高的催化活性和对酶的耐受性。类似地,Akai 等[48]将制备的氧化钒催化剂固定在孔径为3 nm 的介孔二氧化硅(MPS)中得到V-MPS。MPS 有效地将化学催化位点与酶催化位点分隔开,使两种催化剂能够在同一介质中相容。
将有机金属催化剂包埋在蛋白质支架中制备人工金属酶被认为是解决催化剂接触失活的有效方法。金属催化剂被限制在宿主蛋白内部,有效地避免了与外部其他酶的接触。Ward 等[49]将金属复合物[Cp*Ir(Biot-p-L)Cl]包埋到链霉亲和素(Sav)中制备出人工转移氢化酶(ATHase)。ATHase 保留了贵金属的催化活性,并且与天然酶具有良好的相容性。为了证明该方法的通用性,该课题组成功地将ATHase与三种不同的天然酶结合,设计了一系列级联反应。该课题组利用这一策略还制备出了具有原位再生NADH 能力[50]和依赖NAD(P)H[51]的人工金属酶,并应用于不同的级联反应。
然而单独固定某一类催化剂存在一定的隐患,因为固定后的催化剂难免会发生泄漏,泄漏的催化剂同样存在使另一种催化剂失活的可能。将两类催化剂分别固定于不同载体上,不仅可同时提升两种催化剂的稳定性,还可借助载体对催化剂的保护作用消除催化剂泄漏所带来的隐患。理论上,对于分隔催化剂活性中心的效果而言,将两种催化剂分别固定在不同载体上是更优的选择。
金 属 有 机 骨 架(metal-organic frameworks,MOFs)以其可调的孔道结构、高比表面积和大孔隙率等优势近年来受到了研究者们的广泛关注[52-53]。Li 等[54]将Pd 固 定 于MIL-101 中,同 时 还 制 备 了CalB-CLEAs 交联酶聚集体,利用二者开发了一种高效的化学-酶催化体系用于1-苯乙胺的动态动力学拆分。该过程采用微波辐射技术不仅加快了外消旋速率,还降低了反应温度。固定化后的催化剂具有超高的转化率和对映选择性,而且重复使用9次后仍可维持很高的活性。Groger 等[55]将化学和生物催化剂分别固定于不同的高吸水性树脂上,用于合成1,3-二醇。
1.2.2 分隔式集成催化剂 集成催化剂一般分为两类,一类需要载体作为媒介,将酶与金属共同固定于载体上;另一类则无须载体直接通过生物结合的方式将金属连接在酶表面的氨基酸残基上。无论是生物结合法还是简单无差别的共固定方法都无法满足催化剂空间分隔的目的,因为在制备过程中不同催化剂就已经发生接触失活。将不同的催化剂有差别地固定在同一载体的不同位置上制备分隔式集成催化剂,才能有效地杜绝催化剂发生接触失活。分隔式集成催化剂的另一大优势是在避免催化剂活性中心接触的前提下,大大缩短了活性中心的距离,从而提高中间产物的局部浓度,加快反应速率并且使反应平衡向产物的方向移动。制备具有空间分隔催化剂能力的先进纳米材料是该策略的前提条件。在过去的几十年中,纳米材料领域的迅速发展使得研究者可以制备出所需的载体并对其可进行精准调控。制备分隔式集成催化剂一般需要将固定化酶和固定化金属的方法结合起来,前者一般有物理吸附、共价交联和包埋法;后者有共价交联和前体原位生长法。
Jiang 等[56]制备了一种以PtPd 双金属为核、介孔多巴胺为壳的核壳结构纳米颗粒。首先,将Pt和Pd的前体同F127 充分地溶解于水中。然后向混合液中滴加多巴胺溶液,多巴胺有足够的还原力将前体还原成金属颗粒同时自聚形成聚多巴胺(PDA)壳层包裹在金属外表面。去除模板后得到PdPt@PDA 纳米颗粒。随后,采用共价结合法将酶固定于PDA 外表面得到分隔式集成催化剂,如图5 所示。多巴胺壳层有效地将酶与双金属核分隔开,避免了催化剂之间的接触失活现象。介孔结构显著地提高了传质和催化剂的利用率,进而加强了级联反应的协同催化能力。该集成催化剂不仅可以实现一系列伯胺的动态动力学拆分(收率高达99%,光学纯度高达98%),还可以将有机磷农药级联降解为低毒的对氨基苯酚。Prasad 等[57]制备了类似的金属核-壳-酶结构的分隔式集成催化剂。以Au纳米颗粒为核、氧化硅为壳层制备Au@mSiO2,将葡萄糖苷酶接枝到载体的外表面用于化学-酶级联催化反应,同样取得了不错的效果。

图5 PdPt@PDA@CalB的制备过程[56]Fig.5 Formation process of PdPt@PDA@CalB [56]
Wu 等[58]将Pd 纳米粒子(NPs)和CalB 分步负载到功能化介孔氧化硅纳米颗粒不同位置。首先,利用前体原位生长法将Pd NPs 固定于MSN 上得到Pd-MSN。为了提高集成催化剂在有机溶剂中的溶解性,改变颗粒的亲疏水性,采用长链烷烃修饰载体,进而得到疏水性的Pd-mMSN。最后通过疏水相互作用将酶固定于载体上得到CalB@Pd@mMSN。该方法不仅为催化剂的循环利用提供了多相载体,而且还避免了催化剂之间的相互失活。通过表面烷基化修饰后,双功能催化剂可以分散在8 种有机溶剂中,因此所制备的催化剂在甲苯中合成己酸苄酯具有优异的催化性能。Qi 等[59]将Pd 原位生长在MOFs 的孔道结构内,随后通过与月桂酸进行配体交换改变其在有机溶剂的分散性,最后将CalB 等酶固定于不同于Pd 的其他位置上。所制备的催化剂在苯甲醛和己酸乙酯合成己酸苄酯的过程中表现出优异的催化活性。
除了单一孔径材料之外,多级孔材料很好地契合了空间分隔催化剂的需求。Yang 等[60]构建了一种多级孔硅基蛋黄壳@壳结构用于空间分隔Pd NPs与CalB。首先,采用前体原位还原法将Pd NPs 固定于氨基修饰的介孔氧化硅纳米微球上得到Pd/NH2-MSN。随后采用有机硅辅助刻蚀技术制备蛋黄壳结构的Pd/NH2-MSN@BTME。通过双向分层方法,微球被具有大孔的介孔氧化硅壳包裹。最后采用吸附法将CalB 固定于大孔介孔孔道中,而不会进入固定Pd NPs 的小孔中。该结构设计不仅实现了两种催化功能在一个纳米反应器中的固定化,而且还实现了不同催化活性位点的空间分隔。
细胞膜作为天然的“载体”,也为制备分隔式集成催化剂提供了一种可行的策略。细胞膜不仅可以为酶提供最佳的内部环境避免其受到包括化学催化剂在内的外界影响,而且利用整个细胞进行催化直接省略了材料制备、分离纯化酶和固定化酶的过程,仅需将化学催化剂固定在细胞表面即可。Lloyd 等[61]通过生物还原的方法将Pd NPs 固定在可过表达重组单胺氧化酶(MAO-N-D5)的大肠杆菌表面,用于合成对映体纯的手性胺类化合物。该研究首次报道了全细胞生物金属集成催化剂用于一锅多步反应,提供了一种可扩展和高度灵活的平台技术。受到该研究的启发,研究者们基于全细胞开发了一系列集成催化剂用于一锅化学-酶级联反应[62-65]。

图6 利用PDMS套管分隔催化剂活性位点实现Wacker氧化和酶还原反应的级联反应[66]Fig.6 Site-isolation of catalysts using a PDMS thimble for the combination of a Wacker oxidation and an enzymatic reduction[66]
1.2.3 膜分隔(membrane-base separation) 膜分隔技术是将两种催化剂分别限制在膜的两侧,选取合适的膜材料限制催化剂的通过,达到催化剂空间分隔的目的。Groger 等[66]在结合CuCl2/PdCl2催化的苯乙烯Wacker 反应和ADH 催化的还原反应时发现铜盐对ADH 有着强烈的抑制作用。聚二甲基硅氧烷(PDMS)膜可以很好地将化学催化与酶催化分隔开,从而避免了化学催化剂对酶的损害。如图6 所示,在PDMS 套管内部进行Wacker 氧化,中间产物在膜内的浓度不断升高,逐渐向膜外扩散,而化学催化剂不会扩散到膜外而是被限制在膜内。扩散到膜外的中间产物被ADH 还原,从而使反应平衡向产物的方向移动,并且ADH 同样限制在膜外。通过该策略在水相中将苯乙烯成功地转化为1-苯乙醇(ee98%~99%)。
Latham 等[18]同样采用PDMS 膜分隔技术将催化剂的活性位点有效地分离,并且将酶制备成交联酶聚集体增大酶的体积,使酶能够被严格地限制在膜的一侧。随后PDMS 膜分隔技术还被用于更多的化学-酶催化体系[67-68]乃至多酶体系[69],证实了该方法的通用性。
1.3 小结
本节介绍了时间和空间分隔等手段解决一锅化学-酶级联反应中常见的催化条件和催化剂本身的不相容问题,部分实例见表1。催化条件主要包括温度、pH、底物浓度和溶剂的不相容。温度与pH 的不相容通常采用连续反应的方式来解决;底物浓度对催化剂的影响需添加溶剂进行稀释;而溶剂带来的问题可借助两相体系和非常规溶剂加以规避。催化剂本身的不相容需要采用制备分隔式集成催化剂和膜分隔的方法将催化剂的活性中心在空间上分隔。这些方法是互补的,在解决实际问题时可选取两种乃至更多的方法以达到最优的效果。
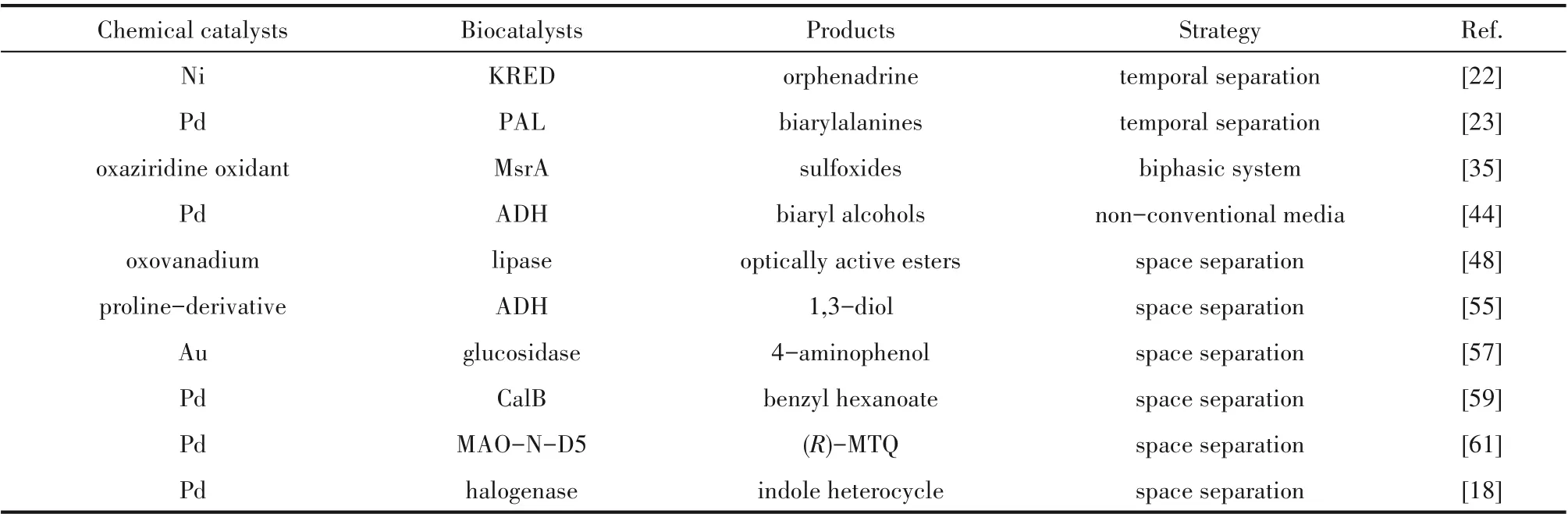
表1 克服化学-酶催化不相容性的实例Table 1 Examples of overcoming chemoenzymatic incompatibility
2 化学-酶级联催化的应用
2.1 动态动力学拆分
胺类和醇类等化合物的动态动力学拆分(DKR)是化学-酶催化反应应用最为广泛的领域之一[70-71]。它由酶催化动力学拆分(KR)和金属催化原位消旋化反应组成,理论产率为100%,克服了传统的动力学拆分产率最高只有50%的局限。对映体纯胺类和醇类化合物是重要的合成原料,广泛应用于农药、食品添加剂、香料和医药等化工产品的生产。
脂肪酶具有优异的对映选择性、通用性和良好的疏水活性,常被用于手性醇和胺的动力学拆分。Jiang 等[56]制备的双金属- 酶集成催化剂PdPt@PDA@CalB 完成了一系列胺衍生物的DKR 反应,并取到了很高的收率与对映选择性,如图7 所示。同样地,Bäckvall 等[72]将Pd 和CalB 共固定于硅基泡沫中用于对伯胺的DKR 反应,与游离的催化剂相比,固定后的催化剂反应效率和转化数更高。Ge等[73]还对比了不同种类Pd-CalB 催化剂用于动态动力学拆分1-苯乙胺的催化能力。
与胺类化合物相比,催化醇类DKR 反应的催化剂种类更为广泛,如V、Al、Ir 等金属配合物,最为常用的是Ru配合物。Bäckvall 等[74]对此进行了大量研究,Ru 基配合物与不同类型的酶结合用于脂肪醇、丙烯醇、氯乙醇、二醇、高烯丙醇和N-杂环1,2-氨基醇的DKR过程。Nolan等[75]报道新型阳离子Ru配合物用于催化仲醇的消旋化(该过程避免了强碱的使用),与脂肪酶结合用于一系列伯醇的拆分。此外,Akai 等发现钒络合物[VO(OSiPh3)3]与各种脂肪酶结合,可用于链状[76]和环状[77]烯丙基伯醇的DKR 反应。随后他们还制备了一种新型的固定化金属催化剂(V-MPD)[48],用于苯甲醇、杂环芳烃和炔丙醇的DKR反应,如图7所示。
除了醇类和胺类的DKR 外,脂肪酶还与碱催化剂或过渡金属催化剂偶联用于合成更复杂的化合物。Ramstrom等[78]通过碱催化的半硫代缩醛形成反应和CalB 催化的分子内酯化反应合成了一系列手性1,3-硫氧杂五元环化合物,最终产物具有良好的转化率和对映体纯度,如图7 所示。该课题组将CalB 与一个动态的多米诺酮加成环化反应进行耦合[79],合成了一系列新的掺杂N、O和S的六元杂环。
2.2 药物中间体的不对称合成

图7 PdPt@PDA[56],V-MPD与CalB[48]和4-甲基吗啉与CalB[78]动态动力学拆分的产物谱Fig.7 Scope of DKR systems involving PdPt@PDA[56],V-MPD and CalB[48],and 4-methylmorpholine and CalB[78]
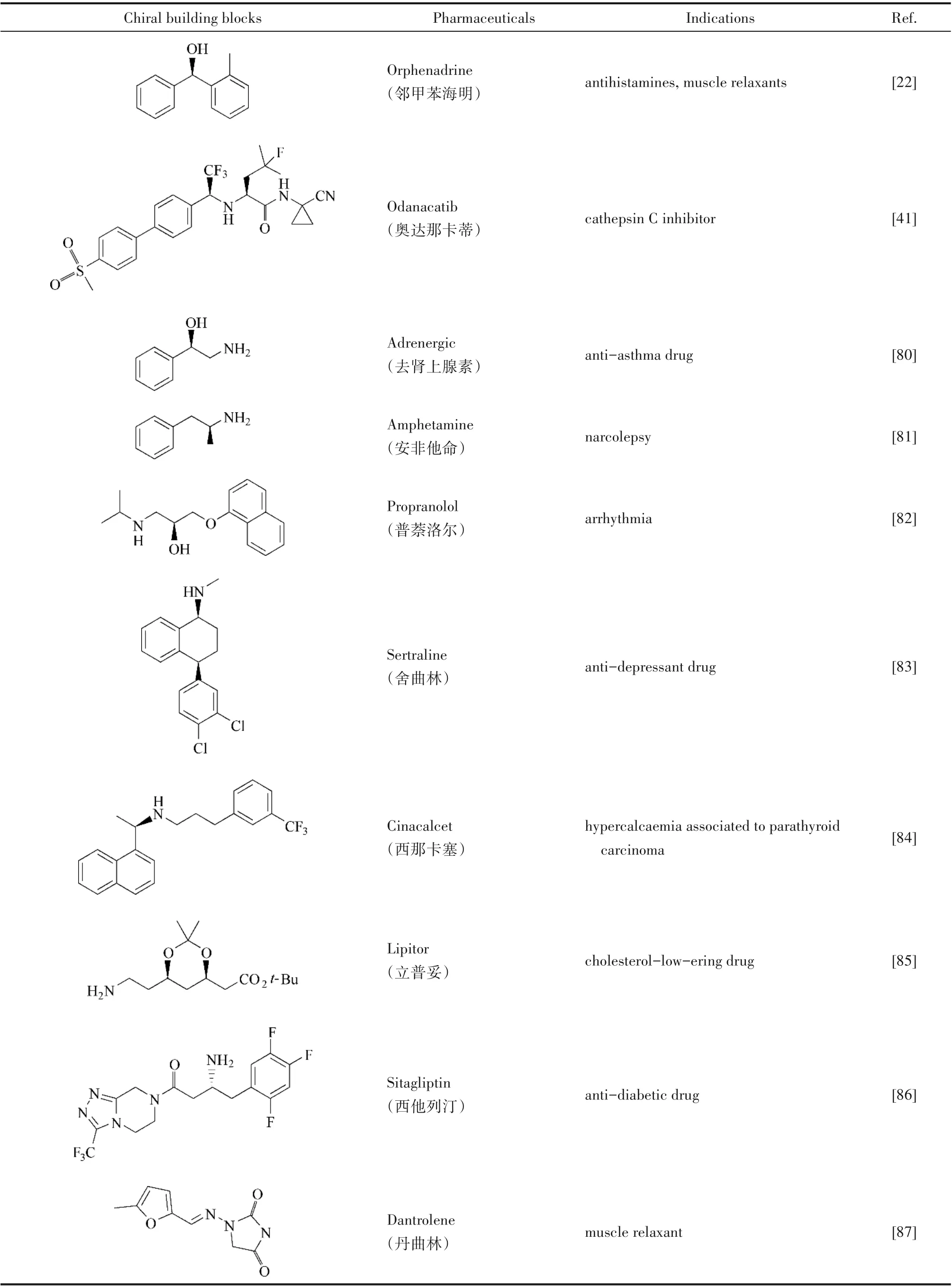
表2 化学-酶催化制备的手性药物与中间体Table 2 Chemoenzymatic catalyzed preparation of chiral drugs and intermediates
目前市售的药物有一半以上是手性化合物,化学-酶级联催化在手性药物中间体的合成领域也得到了广泛应用。化学-酶催化合成药物的典型例子包括西他列汀、辛古拉和立普妥的制备,其中立普妥是迄今为止最为畅销的药物之一。Garg等[22]通过将Ni 催化的Suzuki 交叉偶联反应和KRED 进行偶联,实现了在一锅水介质中将胺类化合物转化成手性醇,结合后续的烷基化反应成功制备了抗阻胺药物奥芬那君。整个反应均在水相中进行,合成的产物不仅形成了新的C—C 键,而且还将其中一个参与反应的C 原子变为手性碳,省略了复杂的逆合成步骤。
Hollmann 等[80]将ADH 催化还原反应和Pd 催化的氢化反应结合,由此制备的2-氨基-1-芳基乙醇衍生物是合成抗炎、抗病毒或抗肿瘤药物的重要中间体。利用该化学-酶催化成功合成的药物活性成分(S)-替巴胺是一种天然的苯甲酰胺衍生物,具有抗病毒(HIV)活性,进一步突出了该反应的合成价值。Gotor-Fernández 等[81]将金属催化的烯丙基苯Wacker-Tsuji 氧化反应与ω-TA 催化的生物还原反应相结合,在水介质中获得了9 种具有光学活性的1-芳基-2-胺衍生物,其转化率高达92%,对映体过量高达99%。该类化合物是合成苯丙胺类手性药物的中间体。Xu 等[82]以环氧水解酶催化消旋的α-萘基缩水甘油醚拆分为关键步骤,通过后续的化学转化合成了具有生物活性的普萘洛尔(一种典型的β 受体阻滞剂)。该药物可用于治疗心绞痛和心律不齐等疾病。
Berglund 等[83]开发出一种制备抗抑郁药舍曲林的化学-酶级联催化方法。KRED 催化外消旋四氢萘酮的生物还原,以出色的光学纯度和非对映选择性(ee>99%,dr>99∶1)合成(S,S)-醇。随后利用次氯酸钠作为氧化剂和2-氮杂金刚烷氮氧化物(AZADO)作为有机催化剂,将生成的(S,S)-醇氧化为对映体(S)-酮,得到舍曲林的重要手性前体。该课题组[84]通过化学-酶法利用ω-TA和KRED设计了两种合成路径,均成功合成了能激活甲状旁腺中的钙受体,从而降低甲状旁腺素(PTH)分泌的西那卡塞。通过化学-酶级联催化合成的手性药物和手性药物中间体还有很多,表2 对其中的一些典型案例进行了总结,并介绍了药物的适应症。
3 结 论
本文重点介绍了克服化学-酶催化过程中催化条件和催化剂不相容的策略,并对化学-酶级联催化反应的应用进行了详细汇总。采用时间分隔的连续反应模式、多相催化和非常规介质解决催化条件的不相容性;制备独立固定化催化剂、分隔式集成催化剂和借助膜分隔技术在空间上分离催化剂活性位点,通过避免催化剂接触失活解决催化剂之间的不相容性。这些方法充当了衔接化学催化与酶催化之间的桥梁。尽管如此,发展更为简单通用的方法仍然十分迫切,这对级联催化体系的发展起着关键性作用。今后,借助酶定向进化等技术对酶进行强化,使酶具有更强的抗逆性和更广的反应性,可以大大增加酶与化学催化剂结合的机会,甚至使酶完全替代化学催化剂,使多酶体系取代化学-酶催化体系。另一方面,化学催化剂也将不断发展,如在水相中的稳定性更好,反应条件更温和,选择性更高等。目前用于化学-酶级联催化的化学催化剂大多数为贵金属,例如用于动态动力学拆分的Ru 和Pd,在实际应用中开发廉价高效且绿色环保的催化剂是今后的工作重点。蛋白质工程、材料科学以及合成生物学等不同研究领域的发展和交叉融合将进一步推动化学催化与生物催化的结合,级联催化的反应类型将不断增加,实用性会更强,而且将越来越多地应用于工业化生产。
