新型冠状病毒膜蛋白的生物信息学分析
2021-01-28赵仁生崔艺璇陈柯瑾祁艳艳蔡正达
赵仁生,周 翔,崔艺璇,陈柯瑾,祁艳艳,胡 琳,袁 燕,蔡正达
(1.云南民族大学 民族医药学院 民族药资源化学国家民委-教育部重点实验室,云南 昆明 650504;2.云南省科学技术院, 云南 昆明 650228)
2019年12月中旬,在武汉市发现了不明原因的肺炎,随后被确认为是由新型冠状病毒引起的肺炎[1].新冠病毒现已成为全球范围内严重的公共卫生事件,对人类的健康和生产造成了极其严重的影响[2].通过基因测序发现,新型冠状病毒属于冠状病毒科、β冠状病毒属;是发现的第3个主要感染人类的冠状病毒,与之前发现SARS-CoV和MERS-CoV存在着明显的差异[3-5].SARS-CoV-2感染人之后有较强的神经毒作用[6],此外雪貂和猫也容易被感染,但对猪、鸡、鸭不易感染[7].
冠状病毒是1种被包被的、单链正义的RNA病毒,其大小在26~32kb之间,是目前发现最大的RNA病毒[8].冠状病毒的主要结构蛋白质有突刺蛋白(Spike protein, S)、膜糖蛋白(Membrane glycoprotein, M)、核衣壳蛋白(nucleocapsid protein, N)、包膜蛋白(Envelop protein, E)组成[9-10].其中M蛋白为跨膜糖蛋白,一般有3~4个跨膜结构域,其N末端的亲水结构域暴露于病毒外表面与细胞质膜中的S蛋白结合,在病毒组装过程中发挥着重要的作用;其C端存在于冠状病毒内表面,与N蛋白相结合,主要作用是维持病毒结构稳定,并与病毒的侵染和释放有关[11-12].研究表明M蛋白在病毒组装、养物质运输和形态发生等方面扮演着十分重要的角色[13].现对SARS-CoV-2结构蛋白的研究主要集中于S蛋白,对M蛋白的研究较少[14-15].因此本文以M蛋白的核苷酸和氨基酸序列为研究对象,利用生物信息学技术和方法研究SARS-CoV-2 M蛋白的结构特征、理化性质和进化,以期为SARS-CoV-2药物开发和预防提供一定的理论参考,也为以后深入研究冠状病毒提供一定的理论支持.
1 材料与方法
1.1 材料
从NCBI(https://www.ncbi.nlm.nih.gov/)官网获得SARS-CoV-2 M蛋白质的氨基酸和核苷酸序列,并下载FASTA文件,用于后续分析.
1.2 方法
利用UCS(http://genome.ucsc.edu/)和Protomer 2.0在线分析软件(http://www.cbs.dtu.dk/services/Promoter/)预测M蛋白质的启动子区;利用EMBOSS在线软件(https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)预测M蛋白质的CpG岛;利用PROMO(http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3)在线分析软件预测M蛋白质转录因子结合位点,参数设置:Selectsprcies选择Only human factors和Only human sites,Maximum matrix dissimilarity rate选择0%;利用PortParam(https://web.expasy.org/protparam/)、ProtScale(https://web.expasy.org/protscale/)、SnapGene_4.3.6软件和SignaIP 5.0(http://www.cbs.dtu.dk/services/SignalP/)在线软件分析M蛋白质的理化性质及信号肽;利用TMHMM-2.0(http://www.cbs.dtu.dk/services/TMHMM/)和PSIPRED在线分析软件(http://bioinf.cs.ucl.ac.uk/psipred/)预测M蛋白质的疏水结构和二级结构;利用Phyre2(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index)预测M蛋白质的三级结构;利用NetPhos3.1(http://www.cbs.dtu.dk/services/NetPhos/)、NetNGlyc1.0(http://www.cbs.dtu.dk/services/NetNGlyc/)和IEDB在线软件(http://www.iedb.org/)分别预测M蛋白质的磷酸化位点、糖基化位点以及B细胞抗原位点;利用PSORTII在线软件(https://www.genscript.com/psort.html)预测M蛋白质的亚细胞定位;利用Blast(https://blast.ncbi.nlm.nih.gov/Blast.cgi)在线软件和MEGA7.0软件对M蛋白质进行进化树分析.若无特别注明,所用软件均使用默认参数进行分析.
2 结果与分析
2.1 SARS-CoV-2 M蛋白质基因特征分析
2.1.1 基因获取及其特征
从NCBI中获取SARS-CoV-2 M蛋白质(Gene ID: 43740571)核苷酸序列,为一条单链正义的RNA序列,其长度为669bp,位于 SARS-CoV-2 RNA链的第 26 523~27 191 位核苷酸之间,编码的蛋白质共有222个核苷酸残基组成.
2.1.2 启动子区分析
通过UCSC在线软件截取M蛋白转录起始位点上游 2 000 bp 的核苷酸序列作为启动子区进行分析.由Protomer 2.0在线软件分析表明,在M蛋白基因序列上游存在2个潜在的启动子区,1个临界预测于 1 800 bp 处,得分为0.570;另1个则位于 1 000 bp 处,得分为1.117,表明在 1 000 bp 处存在启动子区的可能性较大.
2.1.3 M蛋白质甲基化CpG岛分析
Cpglot软件分析结果如图1所示:在观测值/期望值>0.6、G%+C%>0.5、长度大于 100 bp 的条件下,没有发现任何的甲基化岛,表明在M蛋白质的启动子区可能不存在甲基化位点.
2.1.4 转录因子结合位点分析
经SPROMO在线软件分析可知(见表1),在M蛋白质5′基因调控区存在17个潜在的转录因子结合位点.
2.2 SARS-CoV-2 M蛋白一级结构分析
2.2.1 理化性质分析
通过PortParam在线软件分析表明:SARS-CoV-2 M蛋白总共有3 600个原子组成,其分子式为C1165H1823N303O301S8;相对分子质量为 25 147.27 Da,氨基酸种类及占比见表2,酸性氨基酸谷氨酸(Glu)和天冬氨酸(Asp)有13个,碱性氨基酸精氨酸(Arg)和赖氨酸(Lys)共有21个;等电点(isoelectric point)pI为9.51,这与碱性氨基酸含量较高有关.
利用PortParam在线软件分析可得:在水溶液中(280 nm)如果M蛋白的所有Cys都形成二硫键,则其消光系数为 52 160 L/(mol·cm);如果所有的Cys都没有形成二硫键,则其消光系数为 51 910 L/(mol·cm).半衰期:30 h(哺乳动物体外网织红细胞),小于 20 h(酵母),小于 10 h(大肠埃希氏杆菌).不稳定系数为39.14,表明其稳定性良好.由SnapGene_4.3.6软件分析可得:M蛋白在还原态下其吸光率(280 nm,0.1%)为2.06,在氧化态下其吸光率(280 nm,0.1%)为2.07.

图1 M蛋白质甲基化CpG岛预测

表1 M 蛋白5′基因调控区转录因子结合位点预测
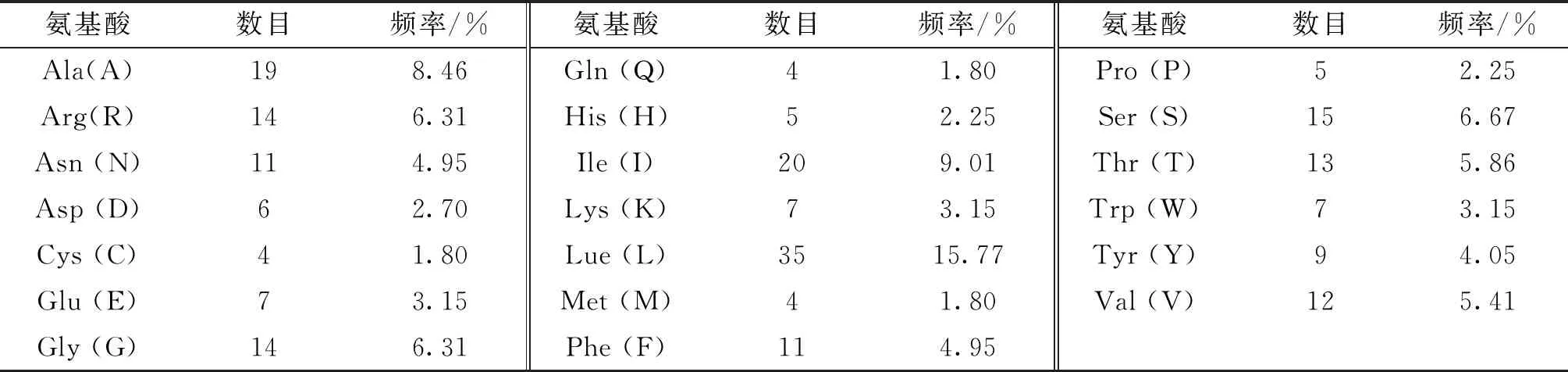
表2 SARS-CoV-2 M蛋白的氨基酸组成信息
2.2.2 疏水性分析
利用ExPSAy在线服务器中的PortParam在线软件分析结果如图2所示:SARS-CoV-2 M蛋白的亲水性总平均值(Grand average of hydropathicity,GRAVY)为0.446(GRAVY在-2与2之间,大于0为疏水性,小于0为亲水性);脂溶指数(Aliphatic index,AI)为120.86,分析结果显示SARS-CoV-2 M蛋白疏水性强,脂溶性高.
2.2.3 M蛋白信号肽分析
经SignaIP 5.0在线软件分析结果显示:M蛋白信号肽值Sec/SPI为 0.000 2(小于0.5),表明在M蛋白中不存在常规的分泌信号肽.
2.3 M蛋白二级结构预测
2.3.1 跨膜结构域预测
PSIPRED预测结果如图3所示,M蛋白存在3个跨膜区:第20~37位氨基酸存在1段由胞外向胞内的跨膜区、第47~67位氨基酸存在一段由胞内向胞外的跨膜区、第77~97位之间存在由胞外向胞内的跨膜区.

图2 M蛋白疏水性分析 图3 M蛋白跨膜区分析
2.3.2 M蛋白质二级结构的预测
PSIPRED在线软件分析结果如图4所示:由A图知,M蛋白存在3个α螺旋、13个无规卷曲和10个β折叠;M蛋白不存在分泌信号肽,这与SignaIP 5.0在线软件分析结果相一致;由B图分析可知,M蛋白中疏水性氨基酸占31.53%、极性氨基酸占22.97%、非极性氨基酸占25.67%,因此可间接反映出M蛋白疏水性较强,这与ProtScale在线软件分析的结果相一致.结果表明M蛋白质二级结构以无规则卷曲和β折叠为主,且为疏水性蛋白.
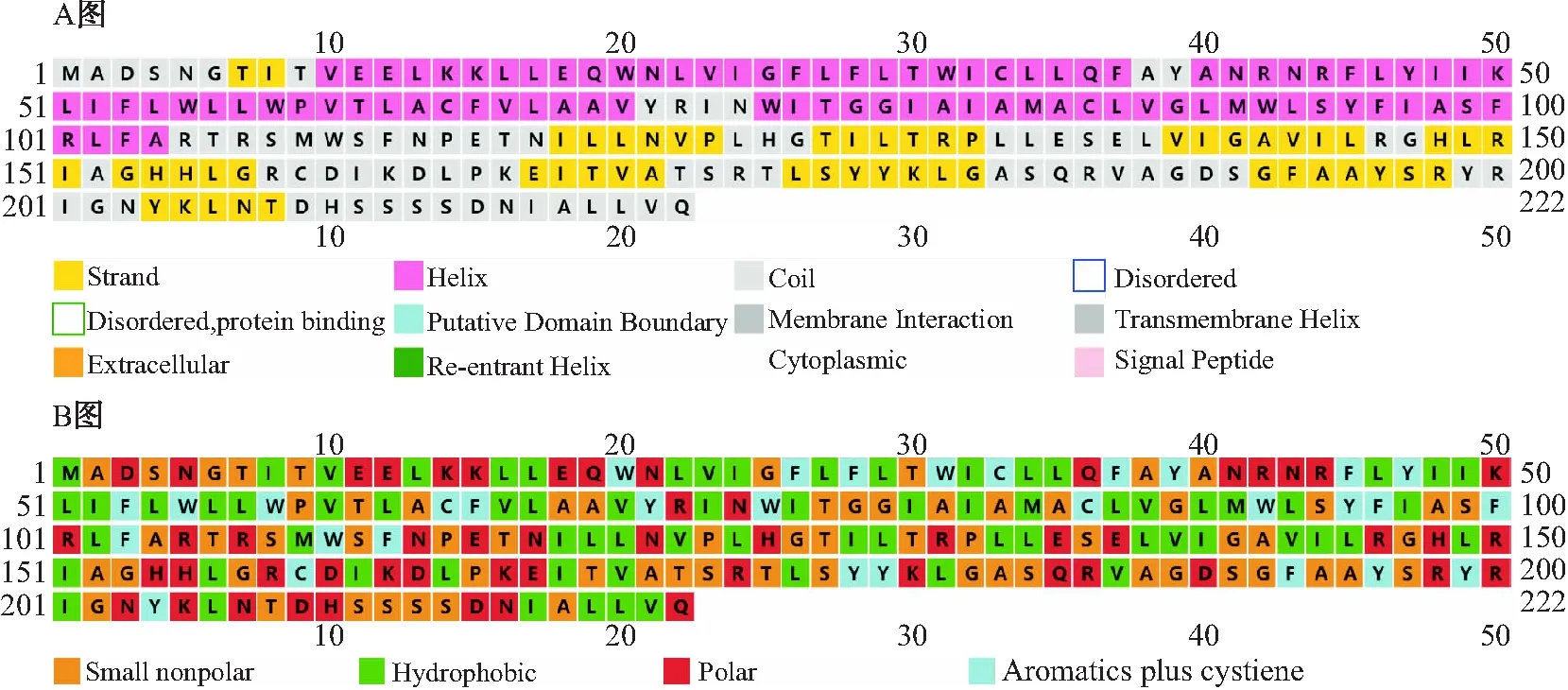
图4 M蛋白二级结构预测
2.3.3 M蛋白质二硫键位置的预测
PSIPRED在线软件分析发现, M蛋白中存在2对二硫键分别位于第32、63、86和159位半胱氨酸残基上,其评分均为8,表明这些位置存在二硫键的可能性较大.
2.4 M 蛋白三级结构分析
由于在PDB数据库中未能找到与SARS-CoV-2 M蛋白质相似性性高(>30%)的模板,因此利用远程同源建模(相似性<30)的方法在Phyre2在线软件中分析M蛋白质的三级结构.以sars-cov-2 orf3a(相似性为25%)模板分析得到了M蛋白质的三级结构,其建模的可信度为58.3%,这提示我们现在对冠状病毒M蛋白质的研究较少.如图5所示,M蛋白的二级结构以无规则卷曲和β折叠为主,2个β折叠结构较长,其余都比较短,这与PSIPRED软件预测结果相一致,此外M蛋白质的三级结构比较松散,这可能与其形成的跨膜结构域和细胞质膜的流动性有关.
2.5 M蛋白质磷酸化和糖基化位点分析
2.5.1 磷酸位点化分析
通过NetPhos3.1软件分析发现(见图6):M蛋白存在37个磷酸化位点,分别为:15个丝氨酸(蓝色)、13个苏氨酸(红色)和9个络氨酸(黑色)磷酸化位点.
2.5.2 糖基化分析
NetNGlyc 1.0 Server在软件分析M蛋白糖基化修饰(见图7),在M蛋白质第5位天冬氨酸残基上存在1个潜在的N-糖基化修饰.

图5 M蛋白三级结构分析

图6 M蛋白质的磷酸化位点预测 图7 M蛋白质糖基化位点分析
2.6 M蛋白质B细胞抗原表位的预测
经IEDB在线软件预测M蛋白的B细胞抗原(见表3),共存在可能的抗原表位有6个,位于N端第5位和第20位之间,长度为16个氨基酸的B细胞抗原表位最强.因此,可以推测M蛋白质主要的B细胞抗原区域可能位于第5位和第20位氨基酸之间.

表3 M蛋白质B细胞抗原表位预测
2.7 M 蛋白亚细胞定位
通过PSORTII在线分析软件预测可得(见表4),当SARS-CoV-2病毒侵染细胞后,其M蛋白在宿主细胞中可能存在的位置为:在细胞的内质网中存在的可能性最高(44.4%),其次是在囊泡中存在的可能性为22.2%,此外,存在于线粒体、细胞质、高尔基体中的可能性均为11.1%.由此可以推测,当SARS-CoV-2病毒感染细胞后,其M蛋白质主要在内质网内进行修饰和加工,然后以囊泡的形式运送至细胞质中形成SARS-CoV-2病毒的细胞膜.

表4 M蛋白质的亚细胞定位分析
2.8 SARS-CoV-2 M蛋白质的进化树分析
通过Blast分析(见图8),SARS-CoV-2膜糖蛋白质的氨基酸序列与SARS coronavirus Tor2和蝙蝠冠状病毒BM48-31 2008膜蛋白氨基酸序列的相似性较高,分别为90.54%和89.35%.为了进一步分析SARS-CoV-2 膜蛋白与其他物种病毒膜蛋白的进化关系,利用MEGA7.0软件绘制了如图8所示的进化树,图中括号里边的数值表示与SARS-CoV-2的蛋白的相似性,通过比较可知,SARS-CoV-2的膜蛋白与SARS coronavirus Tor2和蝙蝠冠状病毒BM48-31BCR2008的膜蛋白的亲缘关系较近,与蝙蝠冠状病毒HKU4和HKU5的亲缘关系较远,但它们都来自于同1个祖先.

图8 SARS-CoV-2 M蛋白质与其他物种的进化分析
3 讨论
利用生物信息学进行大数据分析,有助于更好的预测蛋白质的理化性质、结构与功能,为研究蛋白质提供了不同的角度,同时也为相应的药物研发提供了不同的思路[16].研究发现,M蛋白质作为新冠病毒结构蛋白质,是1条669bp的单链正义RNA序列,共编码222个氨基酸,在M蛋白质基因序列上游1 000bp处存在启动子的可能性较大,在启动子区存在17个转录因子结合位点,不存在甲基化位点,这表明M蛋白质基因可在宿主细胞内大量的表达,为新型冠状病毒的增殖创造了有利的条件,这从基因转录水平反应了M蛋白在SARS-CoV-2中含量高的原因[10],这提示或许可以通过抑制M蛋白的转录,在一定程度上可以抑制SARS-CoV-2在宿主细胞中的增殖.理化性质分析结果显示,M蛋白质中亮氨酸和疏水性氨基酸的含量较高、疏水性强和稳定性良好,这间接表明SARS-CoV-2病毒膜结构比较稳定,为其在不同介质中存活及传播创造了有利的条件,这或许就是疫情反复爆发的原因之一;分析表明:M蛋白质中存在3个跨膜结构域、1个N连接的糖基化修饰位点、不存在外分泌信号肽、M蛋白质主要存在于内质网中,这表明M蛋白主要是在宿主细胞内质网中合成,合成后不会分泌到胞外,直接用于新型冠状病毒的组装[17].
二级结构分析显示,M蛋白主要以无规则卷曲和β折叠为主,在蛋白质中无规则卷曲主要分布于其表面,这样的结构有利于M蛋白在细胞膜中与其他结构蛋白结合,为SARS-CoV-2病毒膜的组装提供了有有利条件.M蛋白质中可能存在37个磷酸化位点,这表明M蛋白质在新冠病毒的组装、增值等过程中发挥着重要的作用[18],通常磷酸化位点与药物靶点有密切的关系,因此这也为有关药物的研发提供了潜在的可能.由于在蛋白质数据库中未能找到与SARS-CoV-2 M蛋白同源性较高的(>30%)的模板,因此采用远程同源建模的方式建立了M蛋白质的三级结构模型,这也反映出现在对冠状病毒M蛋白结构的研究相对不足,而本研究建立的模型可为冠状病毒M蛋白结构的深入研究提供一定的参考.通过预测发现,在M蛋白中存在6个B细胞表位,其中位于N端第5位和第20位之间,长度为16个氨基酸的B细胞抗原表位最强,这提示我们这段序列有可能作为疫苗研发的候选序列.
通过对M蛋白进化树分析表明SARS-CoV-2的M蛋白质与SARS-CoV-Tor和蝙蝠冠状病毒BM48-31 BCR2008具由较高的相似度和同源性,与蝙蝠冠状病毒HKU4和HKU5的亲缘关系较近.研究发现:冠状病毒的发生和传播都与蝙蝠体内的冠状病毒有非常紧密的联系[19],而SARS-CoV-2的M蛋白与蝙蝠的M蛋白具有共同的祖先,且序列具有较高的相似性,这从侧面表明SARS-CoV-2可能来自于蝙蝠.同时进化树分析结果也显示,SARS-CoV-2的M蛋白质与SARS-CoV-Tor序列具有高度的相似性,这表明2002—2003年发生的SARS-CoV与新冠病毒也有联系,SARS-CoV-2可能是冠状病毒在进化过程中由基因变异而产生的.
本文利用生物信息学的方法对SARS-CoV-2 M蛋白的性质、结构和功能进行了分析,为M蛋白的深入研究奠定了基础,为新冠病毒的防治工作提供了一定的研究方向,但还需要更深入的研究和临床实验的验证.
