CRISPR-Cas系统在植物中的研究进展与监管政策
2021-01-27刘肖静王旭静王志兴
刘肖静,王旭静,王志兴
中国农业科学院生物技术研究所, 农业农村部农业转基因生物安全评价(分子)重点实验室, 北京 100081
CRISPR-Cas9技术的原理是在sgRNA的引导下,Cas9蛋白可以对特定的双链DNA进行切割,利用细胞的天然修复机制可以发生碱基的插入、替换和缺失,从而造成移码突变,实现功能基因的鉴定,获得和培育优良性状的农作物。由于该技术操作简单、高效,因而成为科学研究不可或缺的工具。本文简单地回顾了CRISPR-Cas9的作用原理,详细介绍了最新开发的一系列CRISPR变体,归纳总结了CRISPR-Cas系统在植物中的应用、现阶段面临的挑战、基因编辑植物检测方法与各国的监管政策,以期为CRISPR-Cas系统在植物中的应用研究及监管研究提供参考。
1 CRISPR-Cas9系统的作用原理
在细菌的天然免疫系统内首次发现了CRISPR-Cas家族,它的主要功能是对抗外来入侵的病毒及DNA,包括CRISPR基因座和Cas基因两部分。根据Cas的数目和功能将其分为两个大类共6种类型(Ⅰ~Ⅵ)[1],其中第一大类包含有Ⅰ、Ⅲ和Ⅳ型,它们在靶向切割目标序列时需要多个Cas蛋白协同工作;Ⅱ、Ⅴ、Ⅵ型属于第二大类,区别于第一大类,它们只需要单一的Cas蛋白就能够靶向目标序列。在这两类系统中第二类中的Ⅱ型系统因其组成较为简单,仅以一个Cas9蛋白及向导RNA(gRNA)为核心组成,所以是目前使用最广泛的基因编辑工具[2],工作原理如图1所示。
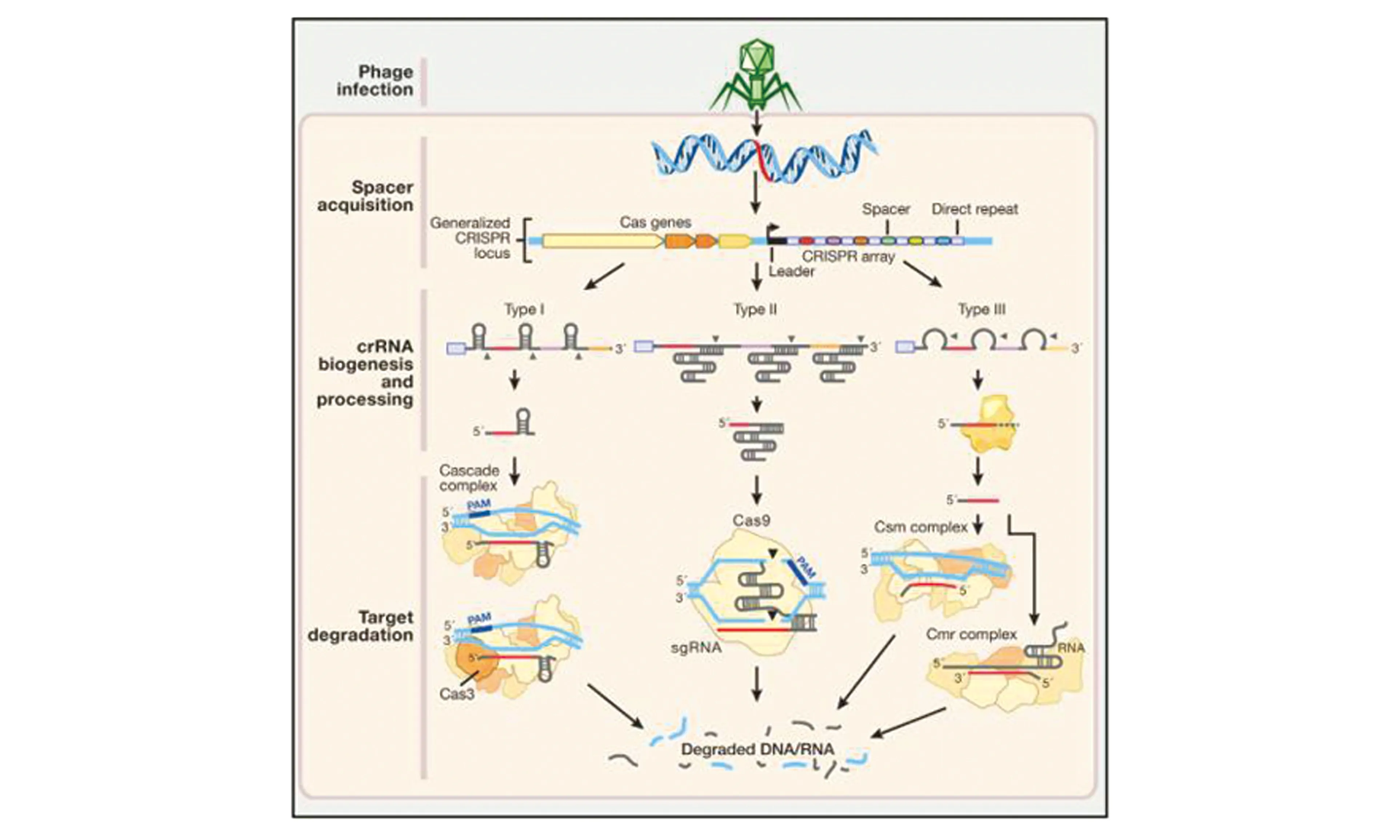
图1 CRISPR-Cas系统的工作原理[3]Fig.1 Natural mechanisms of CRISPR-Cas system[3]
2 新型植物基因编辑器的开发
2.1 CRISPR-Cas12a
Cas12a也被称为Cpf1,是与Cas9最为相似的一个系统。它识别的PAM序列是5′-TTTV-3′,作用原理为利用RuvC-like结构域在crRNA引导下即可切割双链DNA,不需tracrRNA的参与且切割后产生黏性末端,有利于提高基因定点插入的效率;与Cas9相比,Cas12a只需要一个长度为42 nt的crRNA,且它的蛋白也比Cas9的蛋白更小,更加有利于多靶点编辑和小载体系统。CRISPR-Cas12a目前主要用于水稻、烟草、玉米的研究[4-7];该系统和单碱基编辑器成为继Cas9之后被广泛应用的新型基因编辑系统。
2.2 单碱基编辑器(base editing,BE)
单碱基编辑器包含胞嘧啶编辑器(cytosine BE,CBE)和腺嘌呤编辑器(adenine BE,ABE)两种,它是指在不需要供体DNA模板或不产生DSB,甚至不依赖于HDR和NHEJ修复途径的情况下,在靶位点上引起精确且可预测的核苷酸替换;其中CBE编辑器是由大鼠胞苷脱氨酶APOBEC1与Cas9切口酶[nCas9(D10A)]和尿嘧啶糖基酶抑制剂(UGI)融合而成的,在sgRNA的引导下实现了胞嘧啶(C)到尿嘧啶(U)的转换;ABE编辑器是由腺苷脱氨酶与Cas9切口酶融合而成,通过类似于CBE的作用机理实现了腺嘌呤(A)到鸟嘌呤(G)的替换[8]。
目前已在水稻、小麦、玉米、拟南芥等植物中应用了单碱基编辑系统,且在稳定的转基因株系中评估了ABE和CBE的编辑效率和脱靶活性。研究表明,同一个编辑系统在不同靶点位置的编辑效率不同,CBE系统在靶位点处容易发生非特异性的插入和缺失,而在ABE编辑的植物中并未发现非特异性的突变。这表明CBE编辑器比ABE编辑器更加容易产生脱靶,研究发现脱靶现象与Cas蛋白的活性及sgRNA的特异性无关,这可能是由于胞嘧啶脱氨酶的活性造成的[9]。
2.3 xCas9和Cas9-NG
xCas9和Cas9-NG两者都是SpCas9的变体,都在一定程度上扩展了编辑的PAM位点。xCas9可以识别以NG、GAA和GAT为特征的PAM位点,具有较高的DNA特异性和编辑效率;使用xCas9替换Cas9与胞苷脱氨酶或腺苷脱氨酶相融合构建成xCas9单碱基编辑器,对于实现精准编辑目的基因更是意义非凡。Cas9-NG可以识别NG、NAC、NTG、NTT和NCG的PAM位点,并成功用于单基因敲除、多基因敲除、单碱基编辑以及靶基因转录激活调控[10-11]。
2.4 CRISPR-Cas13(C2C2)
CRISPR-Cas13与前面几个CRISPR-Cas变体不同的是Cas13为RNA引导的RNA编辑器,可以特异性靶向切割病毒RNA和真核细胞中的内源RNA。最近研究人员利用失活的Cas13将RESCUE引导到RNA转录本中的目标胞嘧啶碱基上,实现了C到U的转化[12-13],证明了Cas13作为RNA编辑器的潜在广阔应用前景。
2.5 CRISPR-Cas14a
Cas14a能够结合并切割单链DNA(ssDNA),它的蛋白大小约是其他Cas蛋白的一半,且不需要识别PAM序列,是理想的抗植物ssDNA病毒的工具[14]。
2.6 引导编辑(prime editor,PE)
引导编辑是目前最新开发的一种基因编辑方法,该方法可以在不引入双链断裂和供体DNA模板的前提下,实现靶位点的插入、替换、缺失和所有12种类型的点突变。它的组成包括两部分:第一部分为pegRNA,即在我们熟知的sgRNA 3′端加上一段引物结合序列(primer binding site,PBS)和转录模板序列(RT template);第二部分为引导编辑蛋白,由Cas9切口酶(Cas9 nickase仅有切割单链的功能,H840A)和逆转录酶(M-MLV RT)两者融合组成。它的工作原理是Cas9切口酶在pegRNA上的sgRNA序列指导下,切割DNA单链识别位点NGG,pegRNA的引物结合序列(PBS)可以与切割位点前的互补序列识别配对,逆转录酶(M-MLV RT)以pegRNA上的人工设计的模板序列为模板进行逆转录,从而将目标序列直接聚合到切割位点处的DNA链上。通过这个工作原理,可以在无需引入双链断裂和外源DNA供体模板的情况下,有效地产生精确的碱基替换、插入和缺失等突变。
最近该系统已经成功运用在植物领域中,但是它在植物中的编辑效率是极低和不稳定的,因此还需要进一步优化植物载体元件、pegRNA设计的参数、非编辑链切口形成的时间和位置等[15-17]。
3 CRISPR-Cas系统在植物中的应用
3.1 CRISPR-Cas系统在植物基因功能研究中的应用
3.1.1构建植物全基因组突变体库 植物细胞中DSB主要是通过NHEJ途径进行修复,在修复的过程中出现小片段、大片段的缺失或者个别碱基的插入、替换等,从而发生移码突变导致基因的沉默,对基因的功能进行研究。如利用CRISPR-Cas9技术在水稻全基因组范围内构建sgRNA文库创制水稻突变体库对功能基因进行研究,该方法为植物遗传育种提供了新的思路[18]。
3.1.2基因的定点插入和替换 基因的定点插入和替换是通过植物细胞的另一个修复途径HDR实现的,当双链DNA断裂时同时引入一段供体模板,该模板序列两端都携带有与断裂处相同的碱基序列,这时细胞中有可能会发生HDR途径即通过同源重组实现供体序列的精确插入和替换;这种编辑方式有利于研究多基因控制的优良性状,但是由于HDR途径在细胞中发生的概率太低,因此限制了该方式在植物上的应用[19]。
目前提高HDR途径的发生有以下几种方法:①通过抑制NHEJ途径来提高HDR介导的基因靶向效率;②将大量的修复模板供体DNA递送至植物细胞核,粒子轰击可以提供多个供体DNA拷贝。如结合双粒病毒载体构建CRISPR-Cas9系统,通过双粒病毒载体将丰富的供体 DNA 输送到植物细胞中,可获得高达19.4%的靶向碱基替换或插入频率;还有一种方法是使用RNA作为修复模板在植物中实现CRISPR-Cpf1介导的同源重组修复[20],利用转录本RNA作为修复模板,借助于核酶(ribozyme)自切割和 CRISPR-Cpf1系统既切割DNA又切割RNA的特性,通过同源重组修复方式,实现了水稻乙酰乳酸合成酶(OsALS)等位基因的替换,建立了RNA转录本作为修复模板介导的DNA同源重组修复体系。除此以外,有研究报道通过在基因相邻的两个内含子中分别设计一个靶位点,利用NHEJ途径实现基因的定点插入和替换,这个方法的优势在于内含子中双链断裂位点的碱基变异不会影响所在基因的功能。
新开发的单碱基编辑器成为植物中HDR介导碱基精确替换和插入的替代方法[21]。由于这种方法既避免了NHEJ途径修复的随机性,同时也避免了HDR途径修复的低效率性,因而目前已较为广泛地应用在水稻、小麦、拟南芥、玉米等植物中[22-27]。
3.2 作物优良性状的改良和培育
利用CRISPR-Cas系统可以调控基因的表达,从而获得高产、抗病、抗逆等农作物的优良品质。第一种思路是研究者通过对靶基因的启动子活性区域进行编辑,既而改变基因的表达水平与调控模式。例如破坏水稻OsSWEET14启动子中的细菌衍生蛋白结合序列导致对细菌枯萎病的抗性增加;靶向修饰宿主疾病易感基因CsLOB1启动子获得了抗溃疡柑橘品种[28-29];编辑内源基因EPSPS和ALS产生了抗除草剂的水稻;对番茄中多个基因的启动子区顺式调控元件(CRE)的编辑实现了果实大小等重要农艺性状的精准调控[30]。第二种思路为通过dCas9和特定的转录调控结构域结合,形成融合蛋白,从而调控基因的表达。dCas9是Cas9蛋白的一种突变体,即Cas9蛋白的RuvC1和HNA两个核酸酶活性结构域同时发生突变失活,dCas9蛋白失去了内切酶活性,只保留由sgRNA引导进入基因组的能力,这种方法已经在水稻、烟草和拟南芥中得到应用[31-32]。还有一种思路即基因编辑与表观遗传修饰的结合,利用dCas9融合各种表观调控效应器,由sgRNA引导进入目标基因组位点进行特异性的表观调控。如dCas9融合转录因子或转录原件,通过招募转录因子或修饰因子起到调控作用[33-38];或者dCas9和表观遗传修饰酶形成融合蛋白,可以高效地催化DNA和组蛋白的表观修饰,达到了基因的表达调控,目前这种思路在植物中报道很少。
值得注意的是,单碱基编辑器也是利用了缺陷型的Cas9蛋白,但使用单碱基编辑器只产生nick而不产生DSB的nCas9,且它融合的是碱基编辑蛋白胞苷脱氨酶和腺苷脱氨酶;CRISPR介导的表观遗传调控则是使用完全失去内切酶活性的dCas9,它融合的是激活因子和抑制因子等。
同时设计多个不同的sgRNA分别靶向多个基因或者在保守序列区域设计一个sgRNA靶向多个同源基因,这种多靶点编辑方法适用于功能冗余基因和基因家族的研究;例如同时敲除水稻GW2、GW5和TGW6基因,明显提高了水稻的籽粒重量[39];对六倍体普通小麦TaMLO基因的3个同源等位基因同时进行敲除获得了广谱抗白粉病小麦新品种[40]。
4 CRISPR-Cas系统的局限性
尽管CRISPR-Cas系统是一种强大的基因编辑工具,但依然存在很多局限。如:编辑效率的高低、脱靶问题和未知的非预期效应、PAM识别位点的限制、同源重组途径效率低下,以及不同植物的转化问题等。
脱靶效应是指在基因编辑过程中,当sgRNA序列识别出特定序列之外的部分错配而不是靶位点时,就会发生非特异性切割从而产生脱靶效应。脱靶可以导致染色体重排,干扰染色体的稳定性发生未知的非预期效应;还可能导致非靶基因功能活性的丧失,从而导致各种生理或信号异常,影响植物的生长发育。在植物中,CRISPR-Cas系统已在超过30种植物和数百个基因中实现成功编辑[41],脱靶现象的出现对于基因编辑技术的应用带来了很大的不确定性。因此非常有必要对基因组编辑中存在的脱靶问题进行系统分析,进一步提高基因编辑的特异性。
研究表明脱靶情况的出现与靶序列的特异性有关[42~43]。目前,提高基因编辑特异性的方法有三种,第一种方法是提供两个Cas9蛋白,例如两个DNA单链可以被一对Cas9切口酶[nCas9(D10A)]切割。但该方法在编辑位点的选择上存在局限性,且重组载体中含有两个Cas蛋白降低了植物转化的效率。第二种方法是优化改造更多的Cas蛋白变体,如前面所列举的单碱基编辑器(BE)、xCas9等系列Cas蛋白变体,它们扩展了PAM序列的识别位点,扩大了基因组的编辑范围,提高了编辑效率。第三种方法是直接截短sgRNA的长度或者改造sgRNA的结构,已有实验证明截短sgRNA长度可以降低脱靶活性,但是相应的编辑效率也会降低[44-51];研究者通过改造sgRNA二级结构在它的5’末端延伸出1个hairpin发卡结构(hp-sgRNA)将该结构引入到间隔序列(the spacer)上,阻碍脱靶位点R-loop的形成,可有效抑制Cas9在脱靶位点的活性,从而实现CRISPR系统特异性的提高。除此以外,可以利用全基因组测序(WGS)的方法检测脱靶情况,目前已经在水稻和棉花中进行了报道,证实有个别脱靶情况的存在。同时研究人员开发了一系列的脱靶预测工具分析脱靶情况,如:CRISPR-P和CRISPR-GE等,设计sgRNA时应该选择靶点得分高的进行编辑,并在获得编辑的植株后对其潜在预测的脱靶位点进行测序分析。
5 基因编辑植物的检测方法与监管政策
5.1 基因编辑植物的检测方法
利用CRISPR-Cas系统在植物中进行基因编辑后,获得的植株将产生不同的突变。目前基因编辑位点检测方法最常用的有3种:Sanger测序的靶点检测方法、T7E1(T7 endonuclease I)法和高通量二代测序NGS(next generation sequencing)。
5.1.1Sanger测序的靶点检测方法 该方法通过特异性引物扩增编辑位点序列进行测序,测序结果显示正常的通过与野生型靶序列比对得到基因纯合突变类型的植株,如果测序结果显示双峰,首先观察双峰位点是否从切割位点附近开始,然后构建克隆T载体,并挑取多个克隆进行Sanger测序,从而获得多种突变类型的植株;这种方法虽然简便但是只适合少量基因编辑植株的检测,耗时长花费也比较高。
5.1.2T7E1法 该方法是通过特异切割错配分子从而检测突变情况,它可以识别和裂解不匹配的异双链DNA,可以检测到长达12 nt的插入和缺失[52],被认为适用于任何目标序列,但是它的检测灵敏度较低。
5.1.3高通量二代测序NGS 利用该技术可以对编辑的植株进行全基因组测序,获得该植株所有的基因序列信息;目前在分析CRISPR-Cas系统的脱靶情况都是采用这种方法,通过全基因组测序分别检测编辑植株、经过农杆菌转化的野生型植株和野生型植株的基因序列信息,比较获得的SNP、Indel、SNV,从而评估它的编辑和脱靶情况[9]。
5.2 基因编辑作物安全监管政策
CRISPR-Cas系统的优势显而易见,且用途和适用性广泛,在市场商业化方面有广阔的应用前景。由于操作简单、没有外源DNA序列的插入,在监管方面就基因编辑是否以转基因态度来对待,我国尚未出台任何相关规定[39-41];目前不同国家和地区对它的监管持不同态度,还没有形成统一的规定。
5.2.1美国对基因编辑的监管政策 2018年3月28日,美国农业部已经出台政策指出对基因编辑产品不会进行监管。2020年6月,美国农业部给出了支持性的指南,可通过监管获免和未来获免决定监管状况程序来监管基因编辑产品。监管获免是指,植物性状与通过传统育种性状一样或者植物性状已经获得农业部的审批的,不用上市前审批。获得监管获免的植物品种包括3种类型:单碱基的删除、单碱基对的替换、来源于有性亲和的近缘植物的插入——对植物的改变仅包括从有性亲和的近缘植物引入。如果产品没有获得获免,可以写信给APHIS上诉请求审批,如果通过可获得获免,不需要上市前审批,这就是未来获免。后续美国农业部会逐步实施审批指南,以草案形式发布,征求意见[56-58]。
5.2.2日本对基因编辑的监管政策 日本处于一个不欢迎转基因产品的大环境下,日本对基因编辑这种新技术保持谨慎的态度。基因编辑监管的核心问题是基因编辑产品有没有核酸模板的插入,是否会产生除自然或性亲和细胞外的核酸,如果这些插入的DNA来源于自然状态下或者性亲和即为非转基因,反之,最终产品中是否体现该核酸,如没有体现则为非转基因,层层分析,个案分析。
5.2.3澳大利亚对基因编辑的监管政策 澳大利亚基因编辑监管的核心问题也在于基因编辑产品有没有核酸模板的插入,目前已经落实的审议结果是:确定了几种定点核酸技术,SDN1属于自然修复没有核酸模板的插入,属于非转基因;SDN2和ODM有核酸模板的提供,利用同源重组进行修复;SDN3定点插入了一个长片段,有核酸模板的插入;SDN2和SDN3以转基因态度来对待。
5.2.4欧盟对基因编辑的监管政策 欧盟对于转基因有很清晰的定义与立法。欧盟基因编辑监管的关键问题在于是否以转基因的态度来对待基因编辑产品。2018年7月欧洲法院进行了法律解释,即通过诱变获得的生物体属于转基因生物,并且在原则上受到转基因生物法规所规定义务的约束。然而,对于常规在多种应用上使用、且有长期的安全记录的诱变技术,通过其获得的生物体可以免除相关的义务,成员国可与欧盟法规一致,无须遵守指令所规定的义务或其他义务[59-61]。
6 展望
CRISPR-Cas系统因其操作简单、高效、成本相对较低,因而成为植物分子生物学中应用最为广泛的技术之一,也是近几年植物中研究的热点方向。基因编辑技术在作物遗传育种、优良性状改良、功能基因研究中都显现了强大的优势,虽然现在开发出了很多新型的CRISPR-Cas变体,进一步提高了基因编辑的特异性和高效性,扩展了PAM序列的识别位点,提高了同源重组修复途径的编辑效率,优化了植物组培技术,但是脱靶情况仍然不能完全避免,这对于科学研究始终是一个不确定的未知因素,因此在应用基因编辑技术的时候必须考虑分析潜在的脱靶位点,研发编辑范围广、特异性高的CRISPR系统,进一步开发和优化编辑位点检测方法仍然是科研工作者面临的重大挑战和今后突破的方向。
