基于GWAS 数据库鉴定与食管癌相关的易感基因*
2021-01-26史加海
姜 赟,史加海
(1 南通大学附属瑞慈医院胸心外科,南通 226010;2 南通大学附属医院胸心外科)
食管癌是发生在食管上皮组织的恶性肿瘤,是人类常见的消化道恶性肿瘤之一,其侵袭性较高、预后较差。食管癌的患病率在世界各地分布不均,据WHO 报道,全世界约50%的食管癌发生在中国[1]。由于食管癌早期诊断不易,50%以上的患者就诊时已处于中晚期。该病自然病程仅6~8 个月,诊断后的总5 年生存率仅12.8%[2-3]。上皮来源的食管癌主要有两种病理类型:食管鳞状细胞癌和食管腺癌,其中食管鳞状细胞癌是我国食管癌的主要组织学类型,占90%~95%。食管癌的易感性受环境和遗传因素的双重影响,涉及到食管癌发生过程中的危险因素和机制目前尚不清楚。
全基因组关联研究(genome-wide association studies,GWAS)作为研究疾病遗传因素的重要方法,利用芯片通过对单核苷酸多态性(single nucleotide polymorphism,SNP)位点进行检测,发现了与数百种疾病特征相关的变异[4]。2012 年,我国研究者[5]发现rs1050631和rs7242481 与汉族食管鳞癌患者的生存相关联。2016 年发表的一项研究在64 例中国食管癌患者的样本中检测了45 个基因的737 个位点,发现主要的突变基因有TP53、PIK3CA、FBXW7 和KRAS[6]。然而,GWAS 研究中找到与疾病或表型相关的SNPs 大部分位于非编码区[7],通过SNPs 解释其在疾病发病发生机制中的生物学功能时存在难度。DNA 元素百科全书协会研究[8]表明,非编码区的SNPs 也含有调控元件,可间接调控近端或远端基因的表达。全基因组表达定量性状位点(genome-wide expression quantitative trait locus,eQTL)分析是研究SNP 对基因表达影响的一种典型且有效的统计方法[9-10]。eQTL 分析通过测量基因型和基因表达水平数据,使用关联分析或连锁分析寻找基因型和基因表达水平之间是否存在关联[10-11]。
挖掘已发现的与疾病相关的SNP 功能是后GWAS 时代的主要任务和挑战之一。有必要进一步研究这些GWAS 研究中发现的SNP 的作用靶点,以明确它们在疾病发生中的作用机制。因此,本研究通过搜索GWAS 数据库中与食管癌相关的SNPs,利用eQTL 数据库对SNPs 进行靶基因预测,并在食管癌肿瘤组织、癌旁肿瘤组织和正常组织中检测这些基因的表达水平,进一步寻找与食管癌相关的遗传因素。
1 材料与方法
1.1 GWAS 数据库下载与食管癌相关的SNPs 及其功能注释 本研究在GWAS Central 和The Phenotype-Genotype Integrator(PheGenI)网站下载与食管癌相关的SNPs,表型检索词为“Esophageal Neoplasms”。使用UCSC Table Browser 对SNPs 进行相应的功能注释。利用UCSC Table Browser 可获得某些特定的基因或整条染色体的DNA 序列信息和注释信息,并可用文本形式来获取存储在Genome Browser 数据库中的基因组汇编和注释数据[12]。
1.2 eQTL 数据库查找SNPs 调控的基因 通过搜索应用4 个在线eQTL 数据库:https://genenetwork.nl/bloodeqtlbrowser/,http://bioinfo.life.hust.edu.cn/PancanQTL/,http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/和https://www.hsph.harvard.edu/liming-liang/software/eqtl/,筛选SNPs 所作用的具有eQTL 效应基因。
1.3 研究对象 收集南通大学附属瑞慈医院2018—2019 年住院期间行食管癌切除术切除食管鳞状细胞癌组织标本的患者。排除标准:(1)慢性肝肾功能不全的患者;(2)术前曾进行放疗或化疗的患者;(3)有其他部位恶性肿瘤的患者。收集研究对象的一般人口学信息,吸烟史、饮酒史、病史和家族史资料,体格检查资料,实验室检查资料,TNM 分期,临床病理资料,临床诊断治疗信息等。
1.4 RNA 提取,反转录cDNA 后进行逆转录实时定量聚合酶链式反应(reverse transcription-real-time quantitative polymerase chain reaction,RT-PCR)检测术中另保留一份患者的肿瘤组织、癌旁组织和食管远端正常黏膜组织,用去RNA 的无核酶水清洗,剪成<0.5 cm 的薄片后放入离心管,经研磨后得到破碎的组织原液。加入1 mL Trizol 充分吹打裂解,采用苯酚氯仿法提取细胞RNA,并反转录获得cDNA。按照引物设计原则设计待测基因的PCR 引物,见表1。

表1 待测基因引物序列
将cDNA 加入无核酶水稀释5 倍,取出1 μL,分别加入0.5 μL 上、下游引物,10 μL 2×GoTaq qPCR Master Mix 和8 μL 无核酶水,配置成总体积为20 μL的RT-PCR 反应体系,采用GAPDH 作为内参基因,每组设置3 个平行孔,采用ABI QuantStudioTM6 Flex全功能实时荧光定量PCR 系统的标准程序进行RTPCR。反应程序:94 ℃预变性30 s;94 ℃变性5 s,55 ℃退火15 s,共40 个循环。
1.5 统计学方法 采用SPSS 19.0 统计软件对收集的资料进行数据分析。计量资料以表示,符合正态分布的计量资料组间比较采用t 检验,不符合正态分布的计量资料采用非参数检验。计数资料采以n(%)表示,组间比较采用χ2检验或Fisher 确切概率法。对RT-PCR 差异表达基因的计算,先计算△Ct值:△Ct=Ct(检测基因)-Ct(内参基因);再计算△△Ct,△△Ct=△Ct(肿瘤组织/癌旁组织)-△Ct(正常组织),采用2-△△CT方法计算基因表达的差异倍数。P<0.05为差异有统计学意义。
2 结 果
2.1 数据库中发现与食管癌相关的SNPs 在数据库中共发现40 个SNPs 与食管癌相关,来自7 个以东亚人群为研究对象的GWAS 研究,SNPs 的位置信息和-lgP 值如图1 所示。使用UCSC Table Browser对上述40 个SNPs 进行功能注释,只有2 个SNP 位于编码区,18 个SNPs 位于内含子区域,有4 个SNPs,错义突变1 个SNP 位于基因3′端未转译区域。
2.2 与食管癌相关SNPs 具有eQTL 效应的基因在Bloodeqtlbrowser 数据库中,共发现26 对SNPmRNA,其中22 对为顺式作用eQTL(cis-eQTL),4 对为反式作用eQTL(Trans-eQTL)。在PancanQTL 数据库中共发现19 对SNP-mRNA,其中18 对为ciseQTL,仅1 对为Trans-eQTL。在uchicagoeQTL 数据库发现1 个食管癌相关的SNP,具有cis-eQTL 效应。在Liming-liang/software/eqtl 数据库中发现6 对SNP-mRNA,其中5 对为cis-eQTL,1 对为Trans-eQTL。
采用Cytoscape Version 3.6.1 软件对上述数据库中发现的具有eQTL 效应的SNP 和基因的关联进行可视化呈现,结果如图2 所示。共有20 个SNP 参与了对34 个基因的调控作用,其中包含3 个较为复杂的调控网络,如:rs2239815、rs4822983 和rs738722共同参与调控了卷曲螺旋结构域117 基因(coiled-coil domain containing 117,CCDC117)、细胞周期检测点激酶2(checkpoint kinase 2,CHEK2)和HSCB 基因。此外,Cytoscape 还可视化呈现了多个单一的SNP-基因调控关联。
2.3 验证基因确定 选择在以上eQTL 数据库中得到两次验证的基因进行下一步研究,共发现5 个基因在2 个eQTL 数据库得到一致结果,分别是丝氨酸羧化酶基因(serine racemase,SRR),CCDC117、CHEK2、XBP1 和NOC3 样DNA 复制调节因子(NOC3 like DNA replication regulator,NOC3L)。
2.4 食管癌患者的一般情况 本研究中36 例食管癌患者均为食管鳞状细胞癌,年龄54~78 岁,平均(61.86±9.65)岁。男23 例,女13 例,不同性别的患者间吸烟史和饮酒史差异有统计学意义(P<0.05),年龄、高血压、糖尿病、生化检查资料及TNM 分期上差异均无统计学意义(均P>0.05),见表2。
2.5 食管癌肿瘤组织、癌旁组织和正常组织RT-PCR结果的比较 分别从食管癌肿瘤组织、癌旁组织和正常组织提取RNA,反转录后进行RT-PCR 检测SRR、CCDC117、CHEK2、XBP1 和NOC3L 的表达。结果显示:与正常组织比较,食管癌肿瘤组织中CCDC117(t=6.25,P<0.001,差异倍数=1.90)、CHEK2(t=9.37,P<0.001,差异倍数=3.08)、XBP1(t=10.77,P<0.001,差异倍数=2.67)呈现高表达,NOC3L(t=1.86,P=0.06,差异倍数=1.33)和SRR(t=0.24,P=0.81,差异倍数=0.98)的表达差异无统计学意义。此外,肿瘤组织与癌旁组织在CHEK2(t=2.17,P=0.03)和XBP1(t=1.86,P<0.001)的表达上差异也存在统计学意义,CCDC117、NOC3L和SRR 在肿瘤组织和癌旁组织中表达差异无统计学意义,见图3。
表2 食管癌患者基本资料和不同性别之间临床资料的比较(,n,%)
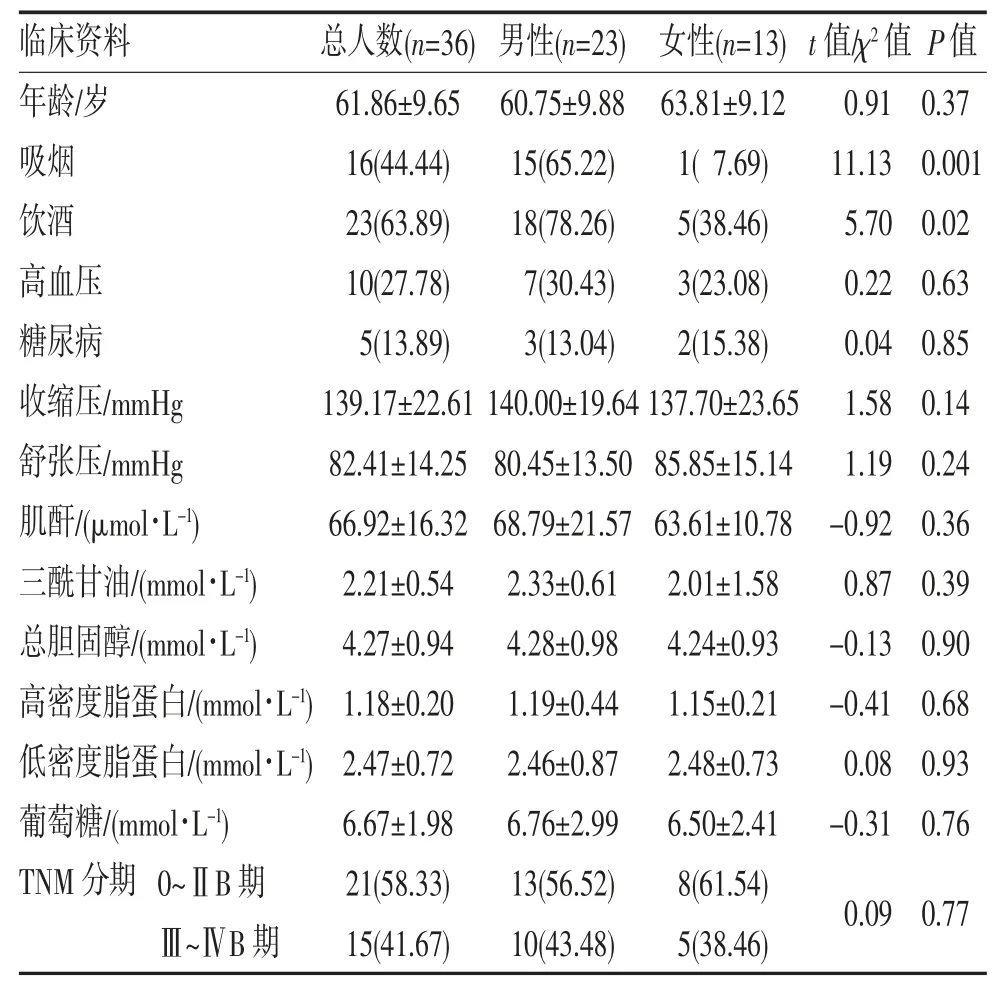
表2 食管癌患者基本资料和不同性别之间临床资料的比较(,n,%)
3 讨 论
食管癌是发生在食管上皮组织的恶性肿瘤,是人类常见的消化道恶性肿瘤之一。众所周知,遗传学上的基因突变是导致肿瘤发生的根本原因,也与食管癌的发生、发展和预后密切相关。GWAS 作为研究疾病遗传因素的重要方法,为研究者探索和识别参与食管癌过程的危险因素或易感基因提供了巨大的机遇。本研究利用GWAS 网站共发现40 个与食管癌相关的SNPs。对这些SNPs 进行功能注释发现大多数SNPs 位于内含子区域。位于内含子本身的SNPs位点可能会通过影响基因的转录水平、剪接(内含子剪接增强子或沉默子)等机制影响基因的功能。
本研究先采用4 个eQTL 在线表达数量性状位点数据库筛选出SNPs 所作用的具有eQTL 效应基因。Bloodeqtlbrowser 对来自5 311 个个体的未转化外周血样本进行了eQTL 分析,并在2 775 个个体中进行重复验证[13]。PancanQTL 综合提供了33 种癌症类型的cis-eQTL 结果(SNPs 影响局部基因表达)和Trans-eQTL 结果(SNPs 影响远处基因表达)[14]。uchicagoeQTL 数据库是来源于芝加哥大学Pritchard实验室和Gilad 实验室的基因调控资源。哈佛大学liming-liang/software/eqtl 数据库分别利用Affymetrix HG U133+2.0 芯片对405 个样本、利用Illumina Human6V1 阵列对550 个样本的淋巴母细胞系的基因表达进行了研究[15]。在4 个eQTL 数据库中共发现20 个SNPs 参与了对34 个基因的调控。
进一步选择在以上eQTL 数据库中得到两次验证的SRR、CCDC117、CHEK2、XBP1 和NOC3L 基因,在食管癌肿瘤组织、癌旁组织和正常组织中进行验证。结果发现食管癌肿瘤组织样本中XBP1 和CHEK2基因的表达比癌旁组织和正常组织高,CCDC117 基因在食管癌肿瘤组织样本中也呈现一定程度的高表达。XBP1 位于人类第22 号染色体上,是亮氨酸拉链蛋白家族的成员之一。XBP1 基因参与人体多种生理功能,在促进细胞生长、分化、促进内质网扩张、抵御氧化应激中都发挥着重要作用[16-17]。研究[18]发现XBP1 在食管鳞状细胞癌肿瘤组织中的表达水平明显高于正常食管组织。CCDC117 是卷曲螺旋结构域家族蛋白成员之一,位于人类第22 号染色体。CCDC家族蛋白成员参与调控多种恶性肿瘤细胞的侵袭、转移。研究[19]发现CCDC33、CCDC34、CCDC67 和CCDC88A 蛋白的表达异常和相关通路改变可明显影响膀胱癌、肺癌、胰腺癌的进展,但目前尚没有对CCDC117 和食管癌进行研究。CHEK2 位于22 号染色体,是一种丝氨酸/苏氨酸激酶,在细胞周期调控、内源性癌基因激活、DNA 双链断裂后的修复等方面起着非常重要的作用[20]。有研究[21]报道了15 例患者的肿瘤和血液标本的全基因组测序结果,发现CHEK2 基因与食管鳞癌有关联。此外,目前还有研究[22-23]发现CHEK2 基因突变与乳腺癌、胰腺癌、前列腺癌等许多恶性肿瘤的遗传易感性有关联。
综上所述,本研究在GWAS 数据库中发现多个与食管癌相关的SNPs 位点,通过eQTL 数据库分析和外周血检测发现这些SNPs 可能通过参与调控基因XBP1、CCDC117 和CHEK2,进而影响食管癌的发生。
