Effect of interaction between loop bases and ions on stability of G-quadruplex DNA*
2021-01-21HanZhenQiao乔汉真YuanYanWu吴园燕YusongTu涂育松andCongMinJi祭聪敏
Han-Zhen Qiao(乔汉真), Yuan-Yan Wu(吴园燕), Yusong Tu(涂育松), and Cong-Min Ji(祭聪敏)
College of Physics Science and Technology,Yangzhou University,Yangzhou 225009,China
G-quadruplexes(GQs)are guanine-rich,non-canonical nucleic acid structures that play fundamental roles in biological processes. The topology of GQs is associated with the sequences and lengths of DNA,the types of linking loops,and the associated metal cations. However,our understanding on the basic physical properties of the formation process and the stability of GQs is rather limited. In this work,we employed ab initio,molecular dynamics(MD),and steered MD(SMD)simulations to study the interaction between loop bases and ions,and the effect on the stability of G-quadruplex DNA,the Drude oscillator model was used in MD and SMD simulations as a computationally efficient manner method for modeling electronic polarization in DNA ion solutions. We observed that the binding energy between DNA bases and ions(K+/Na+)is about the base stacking free energies indicates that there will be a competition among the binding of M+-base,H-bonds between bases,and the base-stacking while ions were bound in loop of GQs. Our SMD simulations indicated that the side loop inclined to form the base stacking while the loop sequence was Thy or Ade,and the cross-link loop upon the G-tetrads was not easy to form the base stacking. The base stacking side loop complex K+ was found to have a good stabilization synergy. Although a stronger interaction was observed to exist between Cyt and K+, such an interaction was unable to promote the stability of the loop with the sequence Cyt.
Keywords: G-quadruplex,loop,DNA base,metal ion
1. Introduction
G-quadruplexes are guanine-rich, non-canonical nucleic acid structures that play fundamental roles in genomic stability and the regulation of gene expression, and also are enriched in promoter sequences of growth regulatory genes,have been a subject of great interest due to the suggested potential in therapeutic applications.[1–4]Various G-quadruplex structures have been observed for human telomeric DNA sequences whose topologies may be associated with some related aspects: the sequences and lengths of telomere DNA,the types of linking loops,and the associated metal cations.[5–9]For example, the formation of G-quadruplex by several G-quartets requires metal ions which dwell in the central of G-quartets are coordinated by several carbonyl oxygen atoms.[10–12]The metal ions related in the stability of G-quadruplex including monovalent cations and divalent cations,among which K+and Na+are widely noted since the most physiologically relevant cations, especially K+, placed between G-quartet layers, accepted as one of the most important factor in the stability of G-quadruplex, but little is known about its process of binding in the human telomeric G-quadruplex.[13–15]On the other hand, stacked G-quartets form a G4 structure, and the intervening sequences are extruded as single-strand loops,although loops are usually small(1–7 nucleotides(nt)),but the sequence and size of loop have a significant effect on the stability of the G-quadruplex.[16]Generally,smaller loops result in more stable G4 structures as do longer G-tracts, and the combination of the type of bounding ions and the location and length of the loops would be expected to lead to a plurality of G-quadruplex structures.[17]
The formation process and stability of G-quadruplexes is complex and difficult to understand.[18,19]Despite a large body of published work on structure and other properties,[20]our understanding of their basic physical properties is rather limited. A large number of papers mentioning G-quadruplexes and many structures that have been deposited in the protein data bank. However, even for short sequences comprising 3–4 G-quartets,it is not known what determines their structures in terms of sequence space, the associated metal cations and the types of linking loops.[21]In this work, we focus on the effect of interaction between base of loop and ions on stability of G-quadruplex DNA.First,the interaction between DNA bases and metal ions was investigated by means of ab initio quantum mechanics energy calculations. And molecular dynamics(MD)simulations were performed to gain insights into the role of metal ions on the stability of GQ with different types of loops. Furthermore, steered MD(SMD)simulations that pulling ion through the channel of G4 structure and loops upon G-quartets were performed to monitor the binding interaction between ion and base in loops based on vary loop basestacking structure, and result in the effect on the stability of loop. Considering the importance of electronic polarization as a contribution to the forces underlying nucleic acid dynamics,the CHARMM36 additive and Drude-2017 polarizable force fields were used in all MD and SMD simulations.
2. Model and method
2.1. Ab initio calculations
The Gaussian 09 suite of program was employed for all ab initio computations.[22]Most calculations were performed at the density functional theory (DFT) level using the polarized split-valence 6-31+G(d,p) basis set.[23]Initial geometries of molecular fragments such as the isolated nucleotide of Adenine(Ade),Cytosine(Cyt),Guanine(Gua),and Thymine(Thy)[24]were built and optimized at the DFT level using a gradient technique. Electron transfers occurring upon DNA base-ion complex formation were evaluated from the variation of the Mulliken gross atomic populations. Although one must be aware of the limitations of the atomic charge partitioning in the Mulliken approximation,[25]we used it here to compare electron charge distributions among different systems. The amount of charge transfer from a DNA-base to the cation was evaluated as the difference of atomic population on the base within the complex and on the isolated base.[26]
2.2. MD system construction
The starting structure for G-quadruplex DNA was taken from the NMR ensemble in PDB entry 1U64.[8]The first of the 8 deposited models was used. The deposited structure lacks bound K+expected in the tetrad core, so two K+ions were added to the GQ structure using the CHARMM program.[27]The positions of the ions were assigned using the average coordinates of guanine base carbonyl oxygen(O6)atoms in consecutive tetrads, yielding symmetric K+coordination. Guanines 2, 8, 12, and 20 comprise tetrad 1, guanines 1, 13, 9,and 21 comprise tetrad 2; and guanines 10, 14, 19, and 22 comprise tetrad 3.Cartoon representation of the structure with bound K+ions named as structure 1 was shown in Fig. 1(a).Two other structures which all the Thy nucleotides in loop of structure 1 were mutated by Ade and Cyt named as structures 2 and 3 respectively, were shown in Figs.1(b)and 1(c). Solvated systems were initially prepared with the additive C36 nucleic acid FF.[28]The GQ was centered in a 70 ˚A×70 ˚A×70 ˚A cubic cell, which was filled with CHARMM-modified TIP3P water[29]and 0.15-mol/L KCl (NaCl), including neutralizing K+(Na+) counterions, which is the typical intracellular KCl concentration in eukaryotes.Standard CHARMM ion parameters were applied to the KCl(NaCl). The energy minimization in NAMD was performed to relax the solvated system.
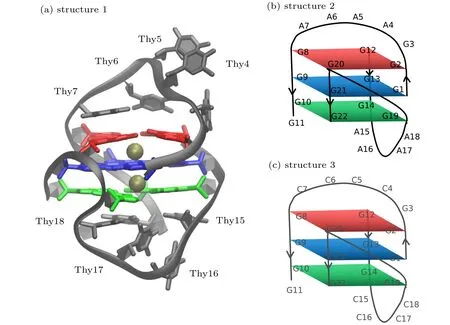
Fig.1. (a)Structure 1: cartoon representation of the NMR structure of human telomeric G-quadruplexes(PDB:1U64)with bound K+ ions(gold). The guanine bases of the GQ core are colored by tetrad(1-red,2-blue,and 3-green);(b)structure 2: schematic structure of structure 1(shown in panel(a))which of all the Thy in loop were mutated by Ade;(c)structure 3: schematic structure of structure 1(shown in panel(a))which of all the Thy in loop were mutated by Cyt.
2.3. MD simulations
Equilibration of the systems was performed in NAMD[30]for 1 ns under an NPT ensemble while all nonhydrogen atoms of the GQ and the bound K+ions were restrained to move and water molecules and bulk KCl ions were free.The NPT ensemble was enforced using the Langevin thermostat method at 298 K, pressure was kept constant at 1 atm(1 atm=1.01325×105Pa) and periodic boundary conditions were applied in all dimensions.The short-range van der Waals forces were smoothly switched to zero from 10 ˚A to 12 ˚A,and the particle mesh Ewald method[31]was used to calculate electrostatic interactions with a real-space cutoff of 12 ˚A and a Fourier grid spacing of 1 ˚A. All bonds containing hydrogen atoms were constrained using the SHAKE algorithm and water molecules were kept rigid with SETTLE, allowing an integration time step of 1 fs.
To prepare the Drude systems:Firstly,each of the three final,equilibrated additive configurations were converted to the Drude polarizable model[32]in CHARMM by adding Drude oscillators to all nonhydrogen atoms in the system. Lone pairs were also added and the TIP3P water molecules were converted to the polarizable SWM4-NDP model.[33]Secondly,the Drude-2017 FF[34]was applied to the GQs and ions,the Drude oscillators were relaxed using steepest descent minimization and adopted-basis Newton–Raphson energy minimization,[35]followed by 1-ns NPT equilibration at 298 K and 1-atm pressure. Extended Lagrangian integration, implemented in NAMD as Langevin dynamics,was used to perform the equilibration simulations.[36]
All unrestrained simulations were performed in NAMD using Drude polarizable FF.Production simulations were carried out for 20 ns each. The same NPT ensemble was maintained as described above, except that for C36 simulations,temperature was maintained using the Andersen thermostat with a collision frequency of 1 ps-1.
2.4. SMD simulations
All SMD simulations were performed in NAMD using Drude polarizable FF and the same NPT ensemble was maintained as described above. Systems sampled from the equilibrium production in which the GQs with different sequence loops were used to SMD simulations in order to study the interaction between ions and loops. In SMD a dummy atom attached by a spring to the K+was pulled up or down(shown in Fig.5(a))with a constant velocity of 0.001 ˚A/fs,while the atom“P”in the phosphate groups of G-quartets of GQs were fixed with harmonic force restraint. The dummy spring had a spring constant 7 (kcal/mol)/˚A2and extension of this spring was used to monitor the force on K+.
3. Results and discussion
3.1. Ab initio calculations on DNA-base and M+-DNA-base complexes
As shown in Fig. 2, electrostatic potential surfaces of four nucleotides were firstly computed at B3LYP/6-31+G(d,p)level,which is different from those of Benz that are symmetrical with the electrostatic potential minima above the ring center as reported earlier.[37]In DNA bases, the presence of heteroatoms strongly disturbs the symmetry of the energy profile observed for benzene, which shifts the electrostatic potential minima relative to the ring center. Even though electrostatic potential above the ring remain favorable with energies within the range of 0.09 a.u. to -0.09 a.u., the electrostatic potential minima are localized above the extracyclic electronegative heteroatoms, namely the nitrogen atoms N1, N3, and N7 of Ade, the two carbonyl oxygen atoms O2 and O4 of Thy, the O6 and N7 atoms of Gua and the O2 and N3 atoms of Cyt.The deepest electrostatic potential minima were observed at the major groove side of Gua and the minor groove side of Cyt. In brief,the electrostatic potential minima were ranked in the order of Ade<Thy<Gua<Cyt,indicating that the strength of the electrostatic interactions is attributed to some extracyclic heteroatoms of Gua and Cyt. Notably, unfavorable electrostatic interaction regions were observed in the vicinity of methyl groups in the DNA bases.
.
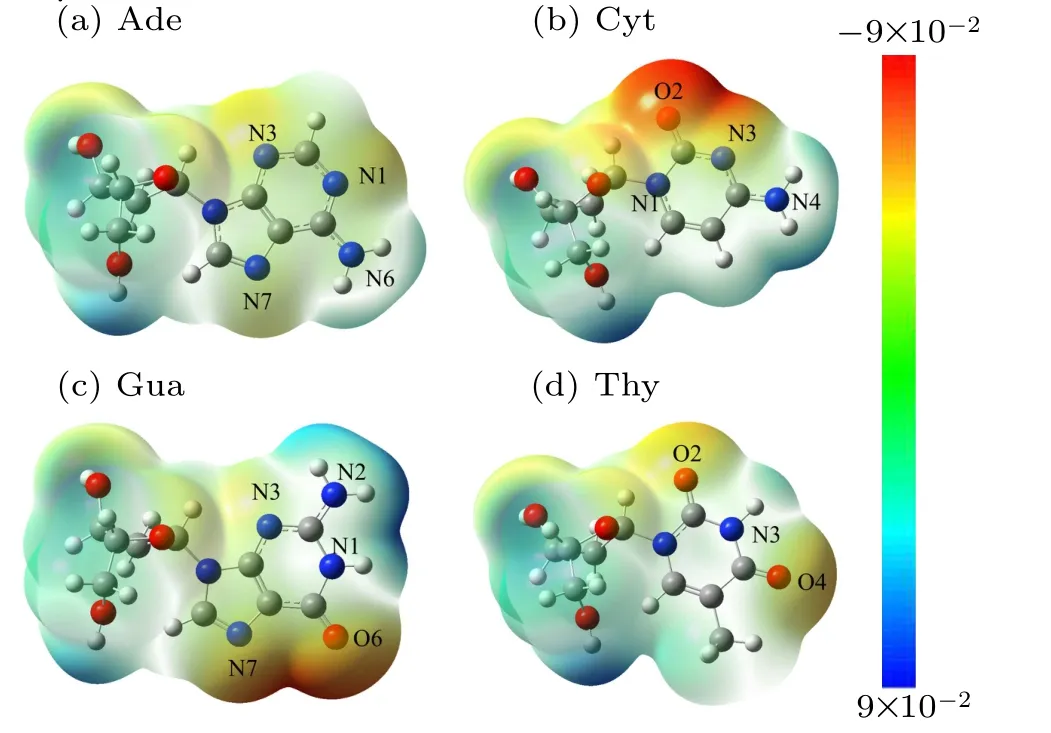
Fig.2. Computed electrostatic potential(ESP)surfaces of DNA bases at the B3LYP/6-31+G(d,p)level of theory electrostatic potential.(Blue is positive;red is negative;red regions are the maximum negative potential). ESP of all the optimized nucleotides were performed using Gaussian 09 software.
The corresponding electrostatic interaction energy between DNA bases and ions(K+/Na+)was revealed to be at the same order. As depicted in Table 1,he binding energy minima ranging between-3.5 kcal/mol to-12 kcal/mol were ranked in the order of Ade<Thy<Gua<Cyt. Whilst the binding energy of Na+–Ade and Na+–Thy were found to be higher than those with K+by about 1 kcal/mol~2 kcal/mol;but the binding energy of Cyt and Gua to K+and Na+were observed to be almost equal. The difference in the electrostatic interaction energy of M+-base may be due to the size difference between K+and Na+. Interestingly,it has been revealed that the binding energies of K+/Na+-DNA bases are attributed to the previously reported base stacking free energies,[38,39]which are larger than H-bond energy, indicating that there is a competition among the binding of M+-base, H-bond between bases the base-stacking while ions are bound in the loop.
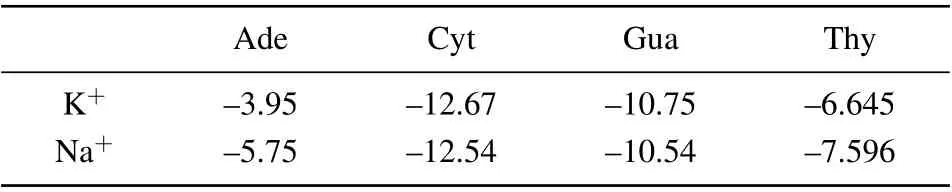
Table 1. Binding energies ΔG (kcal/mol) for M+-DNA bases, calculated at DFT levels of ab initio theory using the 6-31+G(d,p)basis sets.
A detailed analysis of the Mulliken populations shown in Fig.3 indicated that a more pronounced intramolecular reorganization of the electron density occurred while ions are bound to the DNA bases. The ions were firstly placed at positions with maximum negative electrostatic potential around the DNA bases,after which structural optimizations were performed to find the optimized ion-base binding structure. The charge transfer at minimum interaction energy was observed to be about 0.21 e for the nitrogen atoms N1 of K+–Ade,0.22 e for Na+–Ade, 0.16 e for the carbonyl oxygen atoms O2 of K+–Thy, 0.15 e for Na+–Thy. Notably, these values were found to vary for Ade and Thy, and for K+and Na+.Thus,in order to achieve better agreement with QM data and to more accurately simulate the nucleic acids, the application of polarizable force fields is required. The Drude oscillator model used in the MD simulation is a computationally efficient method for the modeling of electronic polarization, in which the auxiliary(Drude)particles are attached to each nonhydrogen atom via a harmonic spring.[40]
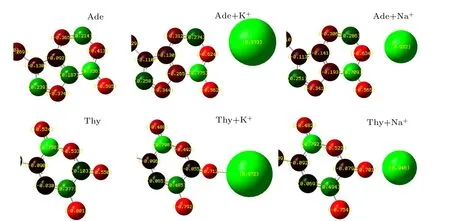
Fig. 3. Charge distribution of nucleotide (Ade and Thy) and charge transfer of K+/Na+-DNA-base complexes computed at DFT/ 6-31+G(d,p) level.Charge distribution of all the optimized K+/Na+-DNA-base complexes were performed using Gaussian 09 software.
3.2. MD simulation on G-quadruplexes in KCl/NaCl solutions
In order to check the stability of the GQs with different loop sequences,equilibrium simulations were conducted during 20 ns.The root mean square fluctuation(RMSF)values for loop atoms of three structures with different loop sequences in 0.15 mol/L KCl/NaCl solutions were calculated. Figure 4(a)shows the RMSF values for the loop atoms of different GQs in 0.15-mol/L KCl. It was observed that the loop of both structures 1 and 2 was considerably more stable than that of structure 3,and that whilst the RMSF of loop atoms in structure 3 showed fluctuations around the equilibrium value of 8 ˚A after 10 ns, the RMSF of loop atoms in structures 1 and 2 showed fluctuations around the equilibrium of value 4 ˚A after 10 ns.Figure 4(b)shows the RMSF values for the loop atoms of different GQs in 0.15 mol/L NaCl. It was observed that the loop of both structures 1 and 2 was slightly more stable than that of structure 3,and that whilst the RMSF of loop atoms in structure 3 had fluctuations around the equilibrium value of 4 ˚A after 10 ns,the RMSF of loop atoms in structures 1 and 2 exhibited fluctuations around the equilibrium value of 3 ˚A after 10 ns.
There is a good correspondence between the structural stability and the base-stacking energies of different loop sequences;for instance,G–A and G–T stacking are more stable than G–C stacking. Furthermore, in KCl solutions, the interaction between Cyt and K+is more stronger than that of Ade–K+and Thy–K+interactions;while in NaCl solutions,the interactions are the same as those in KCl solutions. However,all three structures of the GQs were found to be more stable in NaCl solutions than that in KCl solutions.
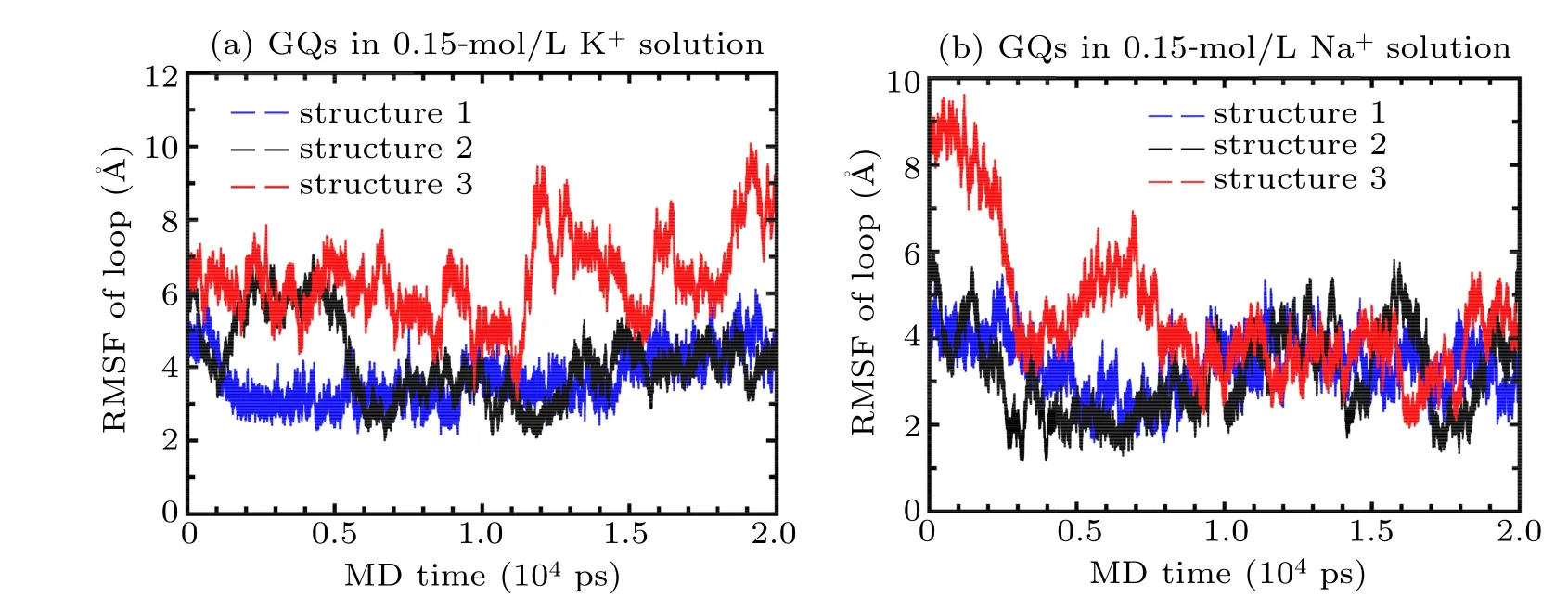
Fig.4. RMSF of loop atoms of G-quadruplexes in(a)0.15-mol/L KCl and(b)0.15-mol/L NaCl solutions(structure 1-bule,structure 2-black,structure 3-red)during 20-ns MD simulation using both CHARMM36 and Drude force field.
3.3. Pulling simulations
SMD simulations were started from the structures obtained in the equilibrium simulations,in which the K+in the central of G-tetrads of structure 1 to 3 were pulled up/down with a velocity of 0.001 ˚A per step while the atom“P”in the phosphate groups of G-quartets of GQs were fixed with harmonic force restraint. The force at the pulling spring as a function of the simulation step is shown in Fig.5.
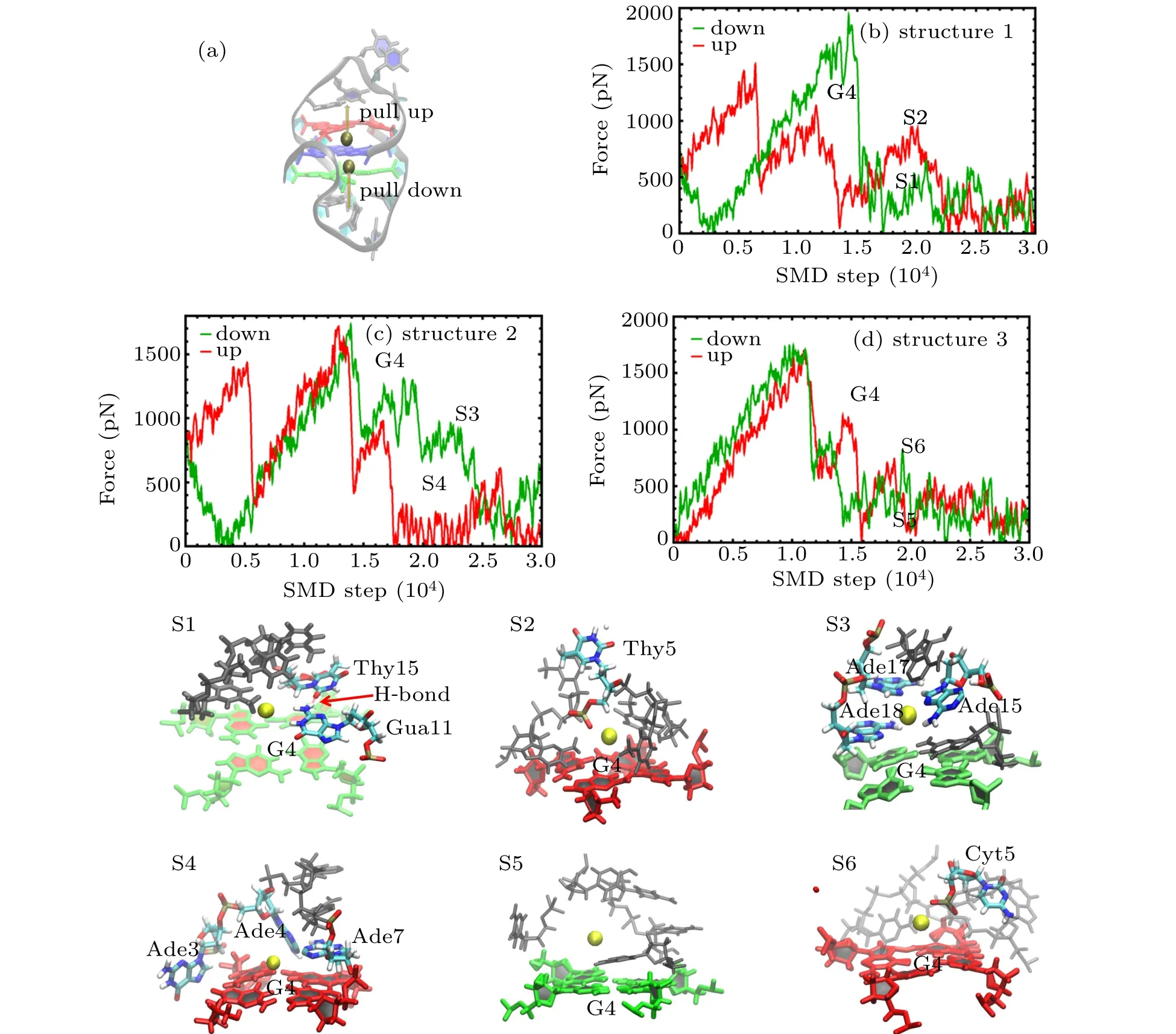
Fig.5. (a)Cartoon representation of the GQ structure with K+ inside the channel showing the pull direction for SMD:pull down(green),pull up(red);(b)–(d)Pulling force on K+inside the channel as a function of time during the pulling simulations for(b)structure 1,(c)structure 2,and(d)structure 3:pull down-green,pull up-red. The G4 in panels(b)–(d)indicates the K+ is pulled through the G-tetrad. The snapshots(S1–S6)correspond to the SMD time marked in panels(b),(c),and(d).
As the pulling process may show certain stochasticity,multiple simulations were performed to show the reproducibility of the pulling process as shown in Appendix A in Fig.A1.The curves displayed a fine position of strong interaction between K+and the loop at high-amplitude force oscillations as a function of the pulling step. Some snapshots at steps marked as S1 to S6 are also presented.
There were differences observed concerning the force values and the position among the different quadruplexes. As shown in Fig.5,ions pulled through the G-tetrad exhibited the highest pulling forces marked as G4,which was similar to the results reported previously.[41,42]Also,there were some force peaks observed while the ions were pulled through the loop.The forces were derived not only from the ion-bases interaction but also from the simultaneous breaking of the hydrogen bonds and the stacking interactions of loop bases induced by the ion pulling. As shown in Fig. 5(b), it was observed that K+pulled up and down through the center of structure 1 with a loop sequence containing Thy, and that there was a small force peak in the pull-down curve after G4. The corresponding snapshot S1 shows that bases Gua11, Thy15, and Thy18 were stacked on the G-tetrad,and that an H-bond was formed between Gua11 and Thy15.The weak interaction between K+and Thy was insufficient to disrupt the stable structure of the loop with several base stacking and H-bonds.There was also a force peak in pull-up curve after G4,and the snapshot S2 that corresponds to this peak shows that the force was derived from the interaction between K+and phosphate group of Thy5,and that there were no base stacking and H-bond formed. As shown in Fig. 5(c), it was found that the force curve of K+pulled up and down through the center of structure 2 with a loop sequence containing Ade,and that there was an obvious force peak in the pull-down curve after G4.The corresponding snapshot S3 shows that that Gua11,Ade15,Ade17,and Thy18 stacked on the G-tetrad by a stronger base staking, and that the peak force was derived from both the volume exclusion and the interaction between the stacking bases and ions. The force curve showing K+being pulled up and down through the center of structure 2 with a loop sequence containing Cyt is depicted in Fig. 5(d). There was a weak peak in both the pull-down and pull-up curve after G4. The pull-down snapshot S5 showed that the weak peak force was derived from the interaction between K+ and Cyt, while the pull-up snapshot S6 showed that the weak peak force was derived from the interaction between K+and the phosphate group of Cyt5.
In all the SMD trajectories of the three GQs studied, it was observed that the side loop in the upper region of G-tetrads was inclined to form the base stacking with sequences containing Thy and Ade, and that cross-linking of the loop on the G-tetrads was incapable of enabling the formation of base stacking easily. In addition, the base stacking side loop complex K+was observed to exhibit a good stabilization synergy corresponding to the RMSD value of structures 1 and 2. For loop sequences containing Cyt,although there was a stronger interaction between Cyt and K+,the interaction was found to be incapable of supporting the loop stability.
4. Conclusion
In this study,we employed ab initio,MD,and SMD simulations to investigate the effect of interaction between loop bases and ions on the stability of GQs.Drude oscillator model,which is a computationally efficient method was used in the MD and SMD simulations for the modeling of electronic polarization in DNA ion solutions. Through extensive calculations and detailed analyses on the changes of loop structures while ions were pulled through different loop sequences, the following major conclusions were obtained.
(i)For DNA bases,the electrostatic potential minima are localized above the extracyclic electronegative heteroatoms.The electrostatic potential minima for bases are ranked as:Ade<Thy<Gua<Cyt. Unfavorable electrostatic interaction regions are observed in the vicinity of the methyl groups of the DNA bases.
(ii)The binding energy minima between DNA bases and ions (K+/Na+) are ranked as: Ade<Thy<Gua<Cyt in the range of -3.5 kcal/mol to -12 kcal/mol. The base stacking free energies indicate that there is a competition among the binding of M+-base,the H-bonds between bases and the basestacking while the ions remain bound in the loop of GQs.
(iii) GQ with Thy or Ade sequence loop is considerably quite more stable than the one with Cyt loop in KCl solutions.
(iv)The side loop is inclined to form base stacking for sequences containing Thy and Ade. Cross-linking of loop on the G-tetrads is unable to enable the formation of base stacking easily. The base stacking side loop complex K+has a good stabilization synergy. For loop sequences containing Cyt, although there is a stronger interaction between Cyt and K+,the interaction is insufficient to stabilize the loop.
Overall,the ionic conditions have an important impact on the topology of the GQs. In this study,we only addressed the stability of GQs in 0.15-mol/L KCl by MD and SMD simulations. Actually,the dynamic process of folding and unfolding of GQs structure plays an important biological function,and the folding topologies of GQ structures critically depend on the sequences and the nature as well as the concentrations of metal ions present in solution,all of which deserve further studies. It has been previously reported that the human telomeric GQ goes through an intermediate state before it is forced to completely unfold, and vice versa. Based on the methods and results in this study, the intermediate states such as Gtriplex should be studied to reveal the folding pathway of the GQ in the future. In addition,the effects of metal ion concentrations on GQs should be addressed. As a preliminary exploration,GQs in 0.5-mol/L KCl solutions were studied by MD,as shown in Fig. A2, the conformation of loop was observed to deviate from the initial conformation in high concentration of metal ion solutions, and the conformational changes was found to be more stable after 10 ns.
Appendix A:Supplementary information
Some experiment results and figures for better understanding the present article are given below.
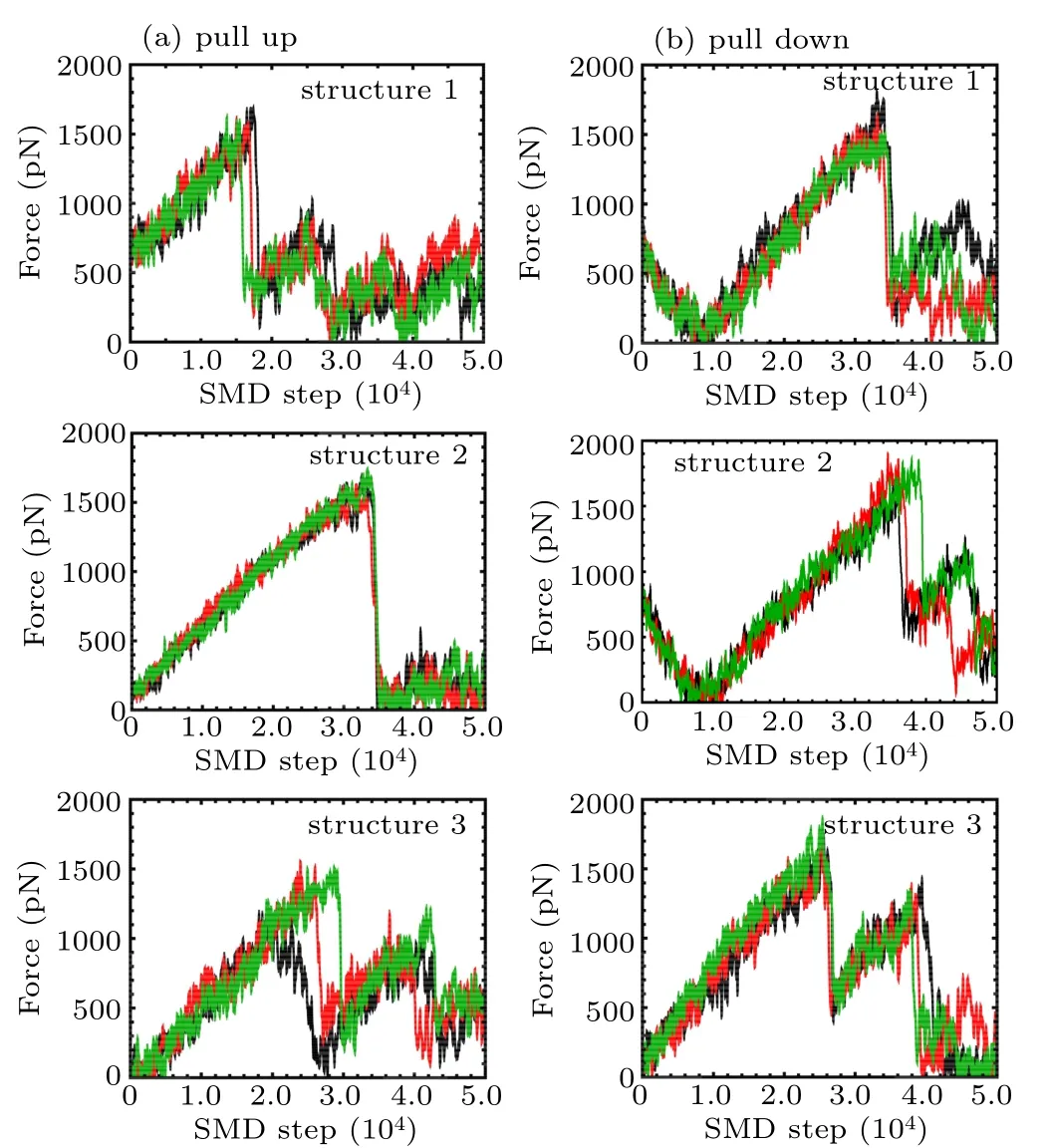
Fig.A1. Pulling force on K+ inside the channel as a function of time during the pulling simulations from multiple pulling trajectories according to Figs.5(b)–5(d), including two pulling directions as shown in Fig.5(a): (a)pull up; (b) pull down for structure 1, structure 2, and structure 3 respectively. Lines with different colors indicate forces derived from multiple pulling trajectories.
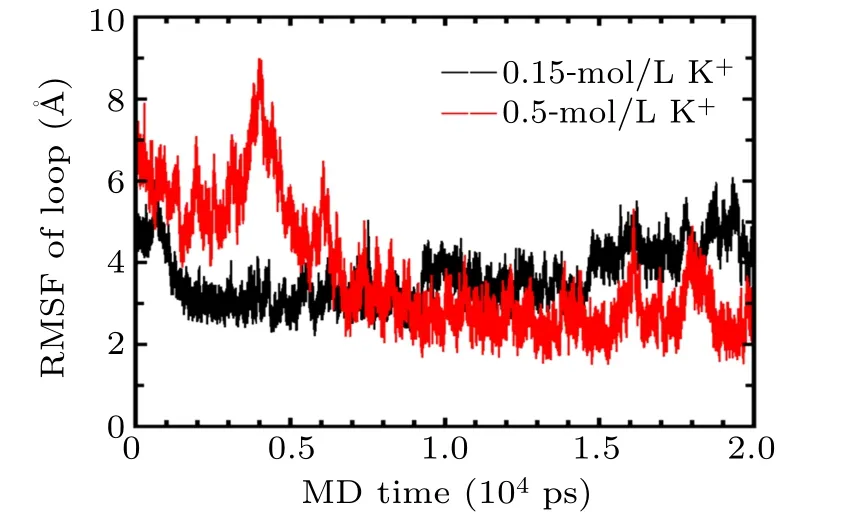
Fig.A2. RMSF of loop atoms of GQs in 0.15-mol/L(black)and 0.5-mol/L KCl (red) solutions during 20-ns MD simulation using both CHARMM36 and Drude force field.
杂志排行

Chinese Physics B的其它文章
- Numerical simulation on ionic wind in circular channels*
- Interaction properties of solitons for a couple of nonlinear evolution equations
- Enhancement of multiatom non-classical correlations and quantum state transfer in atom–cavity–fiber system*
- Protein–protein docking with interface residue restraints*
- Retrieval of multiple scattering contrast from x-ray analyzer-based imaging*
- Numerical research on effect of overlap ratio on thermal-stress behaviors of the high-speed laser cladding coating*