Analysis of long noncoding RNA-associated competing endogenous RNA network in glucagon-like peptide-1 receptor agonist-mediated protection in β cells
2021-01-14
Li-Juan Cui, Lin-Ping Zhi, Huan Xue, Huan-Huan Yang, Xiao-Hua Yang, Yi Zhang, Department of Pharmacology, Basic Medical College, Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
Tao Bai, Yun-Feng Liu, Department of Endocrinology, The First Clinical Hospital of Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
Zhi-Hong Liu, Department of Respiratory Medicine, The First Clinical Hospital of Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
Tao Liu, Department of General Surgery, Shanxi Bethune Hospital, Taiyuan 030006, Shanxi Province, China
Min Zhang, College of Pharmacy, Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
Ya-Ru Niu, Second Clinical Medical College, Shanxi Medical University, Taiyuan 030001,Shanxi Province, China
Abstract BACKGROUND Long noncoding RNAs (lncRNAs) and mRNAs are widely involved in various physiological and pathological processes. The use of glucagon-like peptide-1 receptor agonists (GLP-1RAs) is a novel therapeutic strategy that could promote insulin secretion and decrease the rate of β-cell apoptosis in type 2 diabetes mellitus (T2DM) patients. However, the specific lncRNAs and mRNAs and their functions in these processes have not been fully identified and elucidated.AIM To identify the lncRNAs and mRNAs that are involved in the protective effect of GLP-1RA in β cells, and their roles.METHODS Rat gene microarray was used to screen differentially expressed (DE) lncRNAs and mRNAs in β cells treated with geniposide, a GLP-1RA. Gene Ontology (GO)and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to assess the underlying functions of DE mRNAs. Hub mRNAs were filtered using the STRING database and the Cytoscape plugin,CytoHubba. In order to reveal the regulatory relationship between lncRNAs and hub mRNAs, their co-expression network was constructed based on the Pearson coefficient of DE lncRNAs and mRNAs, and competing endogenous RNA(ceRNA) mechanism was explored through miRanda and TargetScan databases.RESULTS We identified 308 DE lncRNAs and 128 DE mRNAs with a fold change filter of ≥1.5 and P value < 0.05. GO and KEGG pathway enrichment analyses indicated that the most enriched terms were G-protein coupled receptor signaling pathway,inflammatory response, calcium signaling pathway, positive regulation of cell proliferation, and ERK1 and ERK2 cascade. Pomc, Htr2a, and Agtr1a were screened as hub mRNAs using the STRING database and the Cytoscape plugin,CytoHubba. This result was further verified using SwissTargetPrediction tool.Through the co-expression network and competing endogenous (ceRNA)mechanism, we identified seven lncRNAs (NONRATT027738, NONRATT027888,NONRATT030038, etc.) co-expressed with the three hub mRNAs (Pomc, Htr2a, and Agtr1a) based on the Pearson coefficient of the expression levels. These lncRNAs regulated hub mRNA functions by competing with six miRNAs (rno-miR-5132-3p, rno-miR-344g, rno-miR-3075, etc.) via the ceRNA mechanism. Further analysis indicated that lncRNA NONRATT027738 interacts with all the three hub mRNAs,suggesting that it is at a core position within the ceRNA network.CONCLUSION We have identified key lncRNAs and mRNAs, and highlighted here how they interact through the ceRNA mechanism to mediate the protective effect of GLP-1RA in β cells.
Key Words:Type 2 diabetes; β cell; Long noncoding RNA; Competing endogenous RNA;Co-expression analysis; Glucagon-like peptide-1 receptor agonist
INTRODUCTION
The impaired function and diminished mass of β cells in type 2 diabetes mellitus(T2DM) patients lead to insufficient insulin secretion and hyperglycemia[1].Additionally, malfunction, de-differentiation, and apoptosis of β cells are the key characteristics of T2DM[2]. Impaired insulin secretion is a key contributor to chronic hyperglycemia in T2DM patients[2,3]. Hence, a strategy to block β cell apoptosis and restore β cell function is urgently needed. As a class of promising anti-diabetic drugs,glucagon-like peptide-1 receptor agonists (GLP-1RAs) have been shown to potentiate insulin secretion in a glucose-dependent manner, which can decrease blood glucose levels without the risk of hypoglycemia[4-6]. Moreover, studies have demonstrated that GLP-1RAs prevent β cells from premature apoptosis and promote their function[7,8].Recent studies reported that geniposide, a monomer extracted from gardenia, is a novel GLP-1RA with multiple protective effects in human diseases, such as Alzheimer’s disease[9], obesity-related cardiac injury[10], and myocardial ischemia[11]. We and other researchers found that geniposide potentiates insulin secretion, promotes proliferation, and decreases the rate of β cell apoptosis by stimulating the GLP-1 receptor[12-14].
Long noncoding RNAs (lncRNA) are some of the recently studied regulatory molecules[15]. These RNAs are transcripts that are longer than 200 nucleotides and do not code for proteins[16]. Mechanically, lncRNAs exert their regulatory effects through communication with other molecules. Growing evidence demonstrates that lncRNAs regulate mRNA expression by competing with microRNAs (miRNAs)[17,18]. The competition among lncRNAs, miRNAs, and mRNAs was termed the competing endogenous RNA mechanism or “ceRNA mechanism”, which is widely involved in multiple biological processes, including insulin signal transduction that may affect diabetes development[19]. Currently, the mechanisms of the protective effect of GLP-1RA in β cells have been widely investigated, but their potential relationship with mRNAs and lncRNAs is yet to be explored.
We previously demonstrated that geniposide protects pancreatic β cellsviaGLP-1R[13]. In this study, we examined the expression profiles of lncRNAs and mRNAs in INS-1 cells treated with or without geniposideviamicroarray. We further explored,viabiological information analysis, the interaction between lncRNAs and mRNAs, and whether the ceRNA network was involved in their regulatory relationship. We aimed to identify the roles of lncRNAs and mRNAs in mediating the protective effect of GLP-1RA in β cells.
MATERIALS AND METHODS
Cell culture and treatment
Rat pancreatic INS-1 cells were purchased from the National Infrastructure of Cell Line Resource (Identification number: 3111C0001CCC000378). The cells grew irregularly, polygonally, and adherently. Mycoplasma detection was negative. INS-1 cells were grown in high-glucose Dulbecco’s Modified Eagle Medium (DMEM)supplemented with 10% fetal bovine serum (FBS), 100 µg/mL streptomycin, 100µg/mL penicillin, and 50 µmol/L β-mercaptoethanol at 37 °C in an atmosphere containing 5% CO2. INS-1 cells were seeded in 12-well plates to approximately 80%confluence and treated with or without 10 μmol/L geniposide for 24 h. Three technical replicates were performed on each independent sample.
RNA extraction, purification, and quality control
Total RNA was extracted and purified using the RNeasy Mini Kit (Cat#74106,QIAGEN, GmBH, Germany) following the manufacturer’s instructions and checked for an RIN number to inspect RNA integration with an Agilent Bioanalyzer 2100(Agilent Technologies, Santa Clara, CA, United States).
The initial sample of the chip experiment was total RNA that was subjected to quality inspection using a NanoDrop ND-2000 spectrophotometer and Agilent Bioanalyzer 2100 (Agilent Technologies). Then, quality-qualified RNA was subjected to subsequent chip experiments.
Microarray and data analysis
Rat microarray:The Rat microarray Agilent-074571 RAT_LNCRNA_20150413 was developed by Shanghai Bohao Company. The probe information was queried from the GEO database with platform number GPL27603. This microarray was used to profile the lncRNAs and mRNAs. The probe design was based on the latest version of the genome covering core lncRNA and mRNA databases, such as GENCODE V21,Ensembl, UCSC, NONCODE, LNCipedia, and lncRNAdb.
RNA labeling and array hybridization:Total RNA was amplified and labeled with the Low Input Quick Amp Labeling Kit, One-Color (Cat. # 5190-2305, Agilent Technologies), following the manufacturer’s instructions. Labeled cRNAs were purified with the RNeasy Mini Kit (Cat. # 74106, QIAGEN).
Each slide was hybridized using 600 ng Cy3-labeled cRNA and a Gene Expression Hybridization Kit (Cat. # 5188-5242, Agilent Technologies) in a hybridization oven(Cat. # G2545A, Agilent Technologies), according to the manufacturer’s instructions.After 17 h of hybridization, slides were washed in staining dishes (Cat. # 121, Thermo Shandon, Waltham, MA, USA) with the Gene Expression Wash Buffer Kit (Cat. # 5188-5327, Agilent Technologies) according to the manufacturer’s instructions.
Data acquisition and analysis:Slides were scanned with an Agilent Microarray Scanner (Cat. # G2565CA, Agilent Technologies) with default settings: Dye channel,green; scan resolution = 3 μm; PMT 100%; 20 bit. Data were extracted with Feature Extraction software 10.7 (Agilent Technologies). Raw data were normalized using Robust Multichip Average (RMA) algorithm and limma packages in R. Significantly differentially expressed (DE) lncRNAs and mRNAs between the two groups were selected if the fold changes of the threshold values were ≥ 1.5 andPvalue < 0.05.
Gene Ontology analysis
Gene Ontology (GO) analysis can be divided into three parts: Molecular function,biological process, and cellular component, which respectively describe the molecular functions of potential gene products, the biological processes involved, and the cellular environment in which they are located. Enrichment analysis was performedviaDavid 6.8 (https://david.ncifcrf.gov/) database[20,21]. David 6.8 for annotation, visualization,and integrated discovery provides a comprehensive set of functional annotation tools to understand the biological meaning behind the long list of genes. The GO terms obtained in the drawing are arranged in descending order according to the –log10 (Pvalue) of enrichment, and we take the first 10 if there are more than 10 results.
Pathway analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DE genes can enrich the significant pathways and help to find the biological regulatory pathways for significant differences in experimental conditions. The David 6.8 (https://david.ncifcrf.gov/) database also can be used for the enrichment of pathway.The KEGG pathway terms obtained in the drawing are arranged in descending order according to –log10 (Pvalue) of enrichment, and we take the first 10 if there are more than 10 results.
Screening hub mRNAs
Based on the GO and KEGG analyses, a protein-protein interaction (PPI) network was constructed through the STRING (v11.0, https://string-db.org/) database for all DE mRNAs that were enriched in the GO and KEGG terms. STRING is a database of known and predicted protein-protein interactions, including direct (physical) and indirect (functional) associations, which stem from computational prediction,knowledge transfer between organisms, and interactions aggregated from other(primary) databases. Further improvements in version 11.0 include a completely redesigned prediction pipeline for inferring protein-protein associations from coexpression data, an API interface for the R computing environment, and improved statistical analysis for enrichment tests in user-provided networks. The co-expression scores in STRING v11.0 are computed using a revised and improved pipeline, making use of all microarray gene expression experiments deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus[22]. Since our data came from microarray, the protein-protein interactions could be reflected through the STRING database based on the co-expression relationship, and a confidence score ≥ 0.4 was set as the cut-off criterion.
Next, mRNAs in the PPI network were ranked with the Cytoscape(Cytoscape_v3.7.2) plugin CytoHubba, which provides 11 topological analysis methods, including Degree, Edge Percolated Component, Maximum Neighborhood Component, Density of Maximum Neighborhood Component, Maximal Clique Centrality, and six centralities (Bottleneck, EcCentricity, Closeness, Radiality,Betweenness, and Stress), based on the shortest paths. CytoHubba provides a userfriendly interface for exploring important nodes in biological networks[23]. The hub mRNAs were selected from the top threeviaintegrated scores of the 11 algorithms.
Prediction of GLP-1 and geniposide targets
SwissTargetPrediction (http://www.swisstargetprediction.ch/) is an online analysis software for small molecule target prediction, which can predict the target of small molecule compounds based on the principle of molecular similarity. We used this tool to predict the GLP-1 and geniposide targets by converting GLP-1 and geniposide into the standard SMILES format (Canonical SMILES)viathe PubChem database (https://pubchem.ncbi.nlm.nih.gov/), importing the SMILES format file into the SwissTargetPrediction online analysis platform, setting the property to “Rattus norvegicus”, and predicting the targets.
Construction of co-expression network
The Pearson coefficient was calculated based on the normalized chip expression matrix of DE lncRNAs and mRNAs. Molecules were considered with a strong correlation with the filters set atP< 0.05 and |R| > 0.9. These molecules including lncRNAs and hub mRNAs were constructed into a co-expression network.
Construction of ceRNA network
The ceRNA network was constructed based on the relationships among lncRNAs,miRNAs, and mRNAs. It is established that post-transcriptional regulation of mRNAs could be bound by single-stranded miRNAs, and lncRNAs can directly interact by invoking the miRNA sponge to regulate mRNA expression and activity[24]. The specific steps are as follows:
Prediction of hub mRNA-miRNA pairs:The MiRanda (http://www.microrna.org/)and TargetScan (http://www.targetscan.org/vert_71/) databases provide two algorithms for finding genomic targets for miRNAs. The input file included rat miRNA sequences and 3' untranslated region (UTR) sequences of hub mRNAs. The energy and score threshold filters set for MiRanda were -20 kcal/mol and 50,respectively, and the TargetScan binding type filters were set as 8-mer and 7-mer. The intersections of the results from both databases offered the final prediction of hub mRNA-miRNA pairs.
Prediction of lncRNA-miRNA pairs:Rat lncRNA sequences were downloaded from the NONCODE (http://www.noncode.org/index.php) database. The input file included 3' UTR sequences of lncRNAs from the co-expression network and miRNA sequences from last step, which could interact with hub mRNAs. MiRanda and TargetScan were used as described before to screen lncRNA-miRNA pairs. The intersection of the results from both databases offered the final lncRNA-miRNA pairs.
ceRNA network construction:Based on common miRNAs, the ceRNA network was constructed among lncRNAs, miRNAs, and hub mRNAs, indicating that these lncRNAs could co-express with and regulate hub mRNAs through miRNAs.Cytoscape v3.7.1 was used to construct and visualize the ceRNA network.
Statistical analysis
IBM SPSS 25.0 software was used to analyze all statistical data. The random variance modelt-test was employed to identify DE mRNAs and lncRNAs between the control and geniposide-treated groups. Fisher’s exact test was applied for the GO and pathway analyses.Pvalues < 0.05 were considered statistically significant.
RESULTS
Microarray data profile
According to the microarray expression profiling data, a total of 167 upregulated and 141 downregulated lncRNAs were identified in the geniposide-treated group compared with those in the control group with a set filter fold change ≥ 1.5 andPvalue < 0.05. Meanwhile, 28 upregulated and 100 downregulated mRNAs were identified with the same filter settings (Figure 1A and B). This result showed that geniposide treatment induced differential expression of lncRNAs and mRNAs in β cells.
GO and KEGG pathway analyses
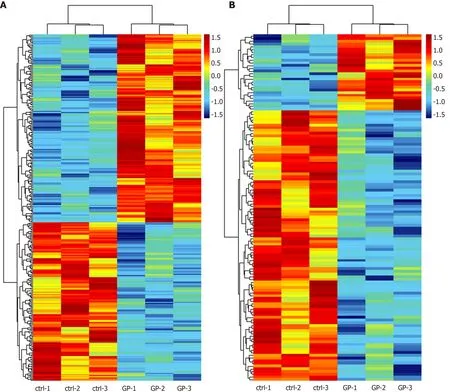
Figure 1 Heatmap of differentially expressed long noncoding RNAs (A) and mRNAs (B).
To investigate the biological function and potential mechanism of DE mRNAs, GO and KEGG pathway enrichment analyses were performedviathe DAVID database(Figure 2). Biological process analysis was mainly enriched in terms of inflammatory response and positive regulation of the ERK1 and ERK2 cascade. Molecular function analysis was primarily enriched in terms of G-protein coupled receptor (GPCR)activity, and the KEGG pathway was mainly enriched in terms of the calcium signaling pathway. Detailed information is presented in Table 1. These findings showed that DE mRNAs participated in biological functions that were closely related to insulin secretion and β cell viability (Figure 2). Next, we investigated the core mRNAs that perform these functions.
Construction of PPI network and screening hub mRNAs
To construct a DE mRNA interaction network, we studied the PPI relationshipviathe STRING database. This network was comprised of DE mRNAs enriched in the GO and KEGG terms, including 120 nodes and 52 edges (Figure 3). To discover the core mRNAs in this network, CytoHubba was used to screen the hub mRNAs. Based on the CytoHubba scores, the top three hub mRNAs,Pomc,Htr2a, andAgtr1a, were identified(Figure 4), indicating that they had more interactions with other mRNAs and participated in various functions and pathways than other hub mRNAs.
Verification of hub mRNA prediction
To confirm the accuracy of the hub mRNA prediction, we queried the GLP-1 and geniposide targets using SwissTargetPrediction tool. One hundred GLP-1 and 80 geniposide targets were predicted with SwissTargetPrediction. Among them, 22common targets were shared by both GLP-1 and geniposide (Table 2). The prediction results showed thatAgtr1andAgtr1bwere among the most common targets.Agtr1andAgtr1b, as well as our hub mRNAAgtr1a, all belong to theAgtrfamily.Additionally,Htr2awas also present in the GLP-1 targets. This was consistent with our mining ofHtr2aandAgtr1aas target mRNAs that mediate the functions of geniposide,confirming the accuracy of our analysis. On the other hand,Pomcwas not found as a GLP-1 or geniposide target per SwissTargetPrediction, but was predicted as the hub mRNA with the highest score by CytoHubba, suggesting thatPomcwas a potential GLP-1 and geniposide target.
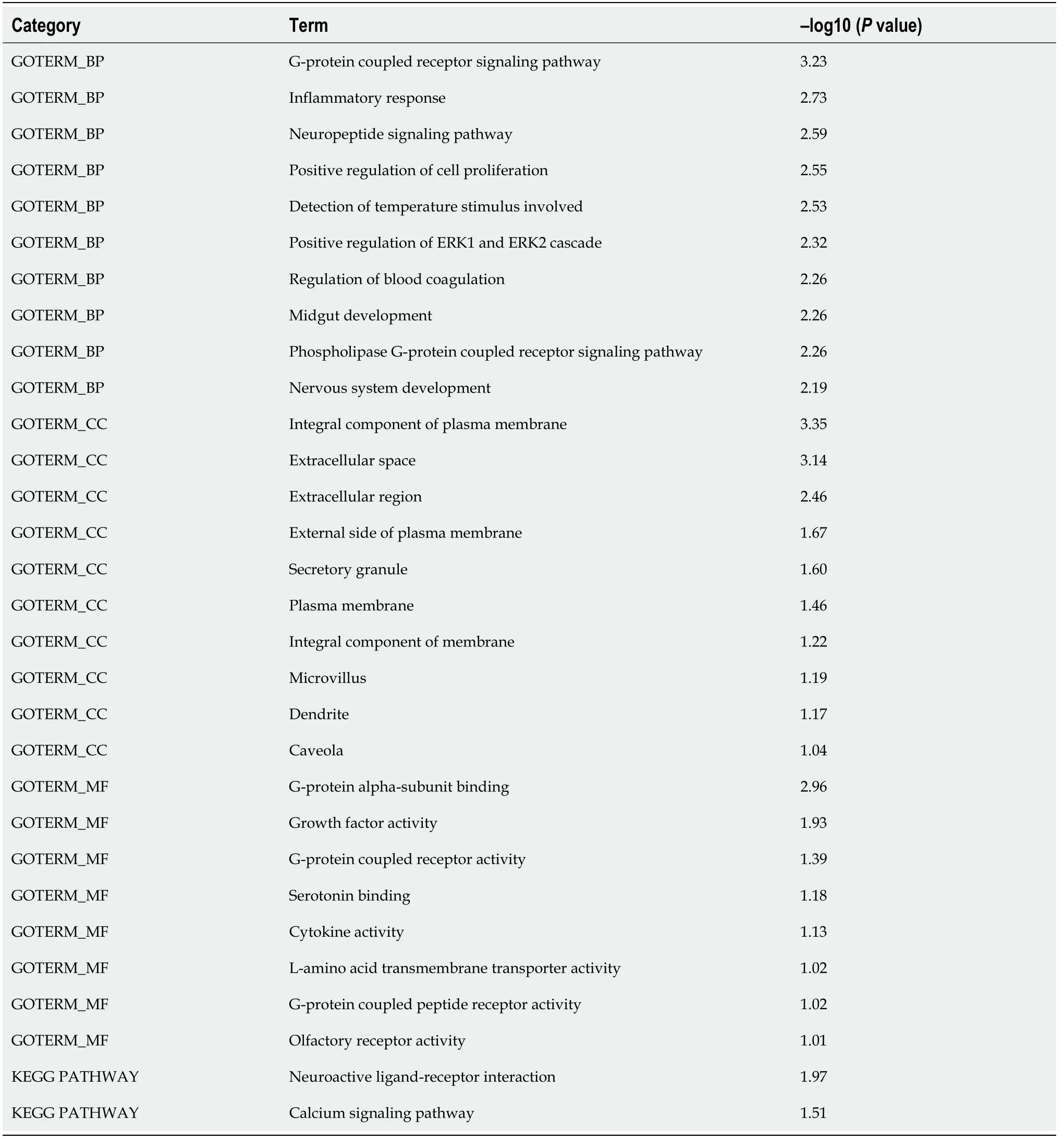
Table 1 Significantly enriched Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway terms of differentially expressed long noncoding RNAs
Co-expression network among hub mRNAs and DE lncRNAs
To explore the key lncRNAs that are closely related to the hub mRNAs, a coexpression network was constructed based on the Pearson coefficient of DE lncRNAs and mRNA expression levels. The number of lncRNAs identified to be co-expressedwithPomc,Htr2a, andAgtr1awas 21 (Figure 5A and 5B). The fold change and the Pearson coefficient of each lncRNA are shown in Table 3.

Table 2 Common molecular targets of glucagon-like peptide-1 and geniposide from SwissTargetPrediction
The expression of the three hub mRNAs (Pomc,Htr2a, andAgtr1a) was downregulated in geniposide-treated INS-1 cells compared to that in untreated cells.Among the 21 co-expressed lncRNAs, 11 (NONRATT002662, NONRATT024273,NONRATT012943, NONRATT027738, NONRATT005090, NONRATT012881,NONRATT005619, NONRATT019513, NONRATT027888, NONRATT006762, and NONRATT030038) were downregulated and positively correlated with hub mRNAs.The correlation between the expression levels of the 11 lncRNAs and hub mRNAs prompted us to explore whether they are functionally related.
Construction of ceRNA network
A ceRNA network was constructed based on the common miRNAs that could bind to the 11 lncRNAs, as well as the three hub mRNAs. Based on the intersection of the prediction results from MiRanda and TargetScan, we obtained 77, 16, and 23 miRNAs that could bind to the 3' UTR ofHtr2a,Pomc, andAgtr1a, respectively. Among these miRNAs, we found that rno-miR-449a-5p could interact with bothHtr2aandPomc,while rno-miR-5132-3p, rno-miR-344g, rno-miR-3075, rno-miR-378a-5p, and rno-miR-874-3p could interact with bothHtr2aandAgtr1a.
Then, we screened lncRNAs that could be bound by these six miRNAs from the 11 co-expressed lncRNAs and found seven such lncRNAs. Based on this analysis, a ceRNA network composed of three hub mRNAs, six miRNAs, and seven lncRNAs was constructed (Figure 6A). This network suggested that lncRNAs(NONRATT002662, NONRATT005090, NONRATT005619, NONRATT019513,NONRATT027738, NONRATT027888, and NONRATT030038) may competitively bind to miRNAs (rno-miR-449a-5p, rno-miR-5132-3p, rno-miR-344g, rno-miR-3075, rnomiR-378a-5p, and rno-miR-874-3p), and thereby affect the expression and function ofhub mRNAs (Pomc,Htr2a, andAgtr1a). Further analysis of the network indicated thatNONRATT027738can regulate all the three hub mRNAs (Pomc,Htr2a, andAgtr1a)through miRNAs (rno-miR-449a-5p, rno-miR-5132-3p, and rno-miR-378a-5p)(Figure 6B).
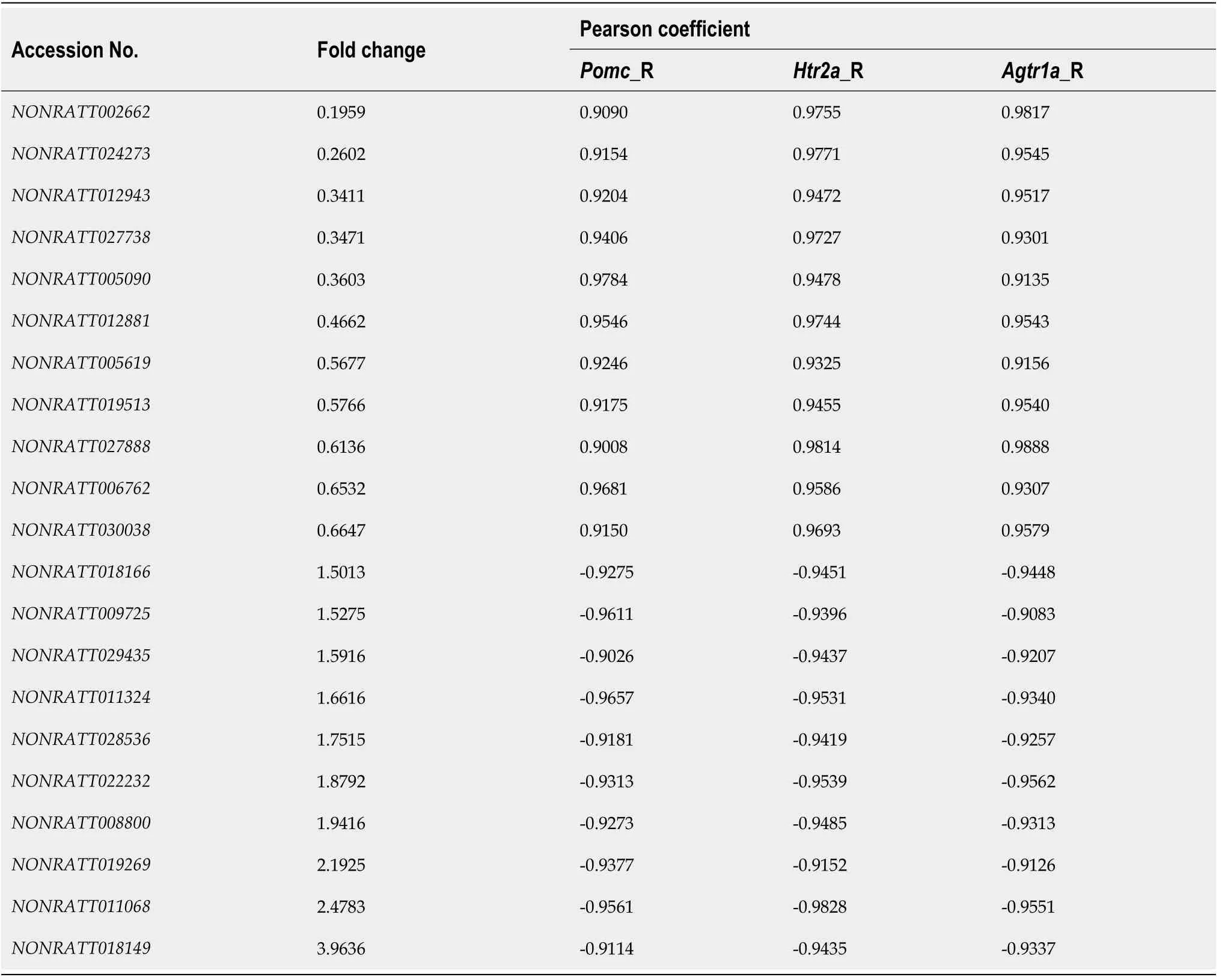
Table 3 Long noncoding RNAs co-expressed with Pomc, Htr2a, and Agtr1a
DISCUSSION
GLP-1RA increases β cell sensitivity to glucose and protects β cells from apoptosis[25].The insulinotropic effects of GLP-1RA are glucose-dependent, posing a low risk for hypoglycemia[26]. To further understand the functions of GLP-1RA in β cells, it is essential to identify the molecular mechanisms involved. Increasing evidence indicates that lncRNAs play key roles in many biological processes, including insulin secretion and cell proliferation by the ceRNA mechanism[27]. In this study, we analyzed the microarray data of INS-1 cells treated with geniposide, which is a GLP-1RA confirmed by plenty of studies[10,13,14]. Our input data were analyzed using bioinformatic tools,including analyses of GO/KEGG pathway, PPI network, co-expression network, and the ceRNA network. With the bioinformatic tools, we identified three hub mRNAs,seven lncRNAs, and six miRNAs that mediated the protective effects of GLP-1RA in β cells through the ceRNA mechanism.
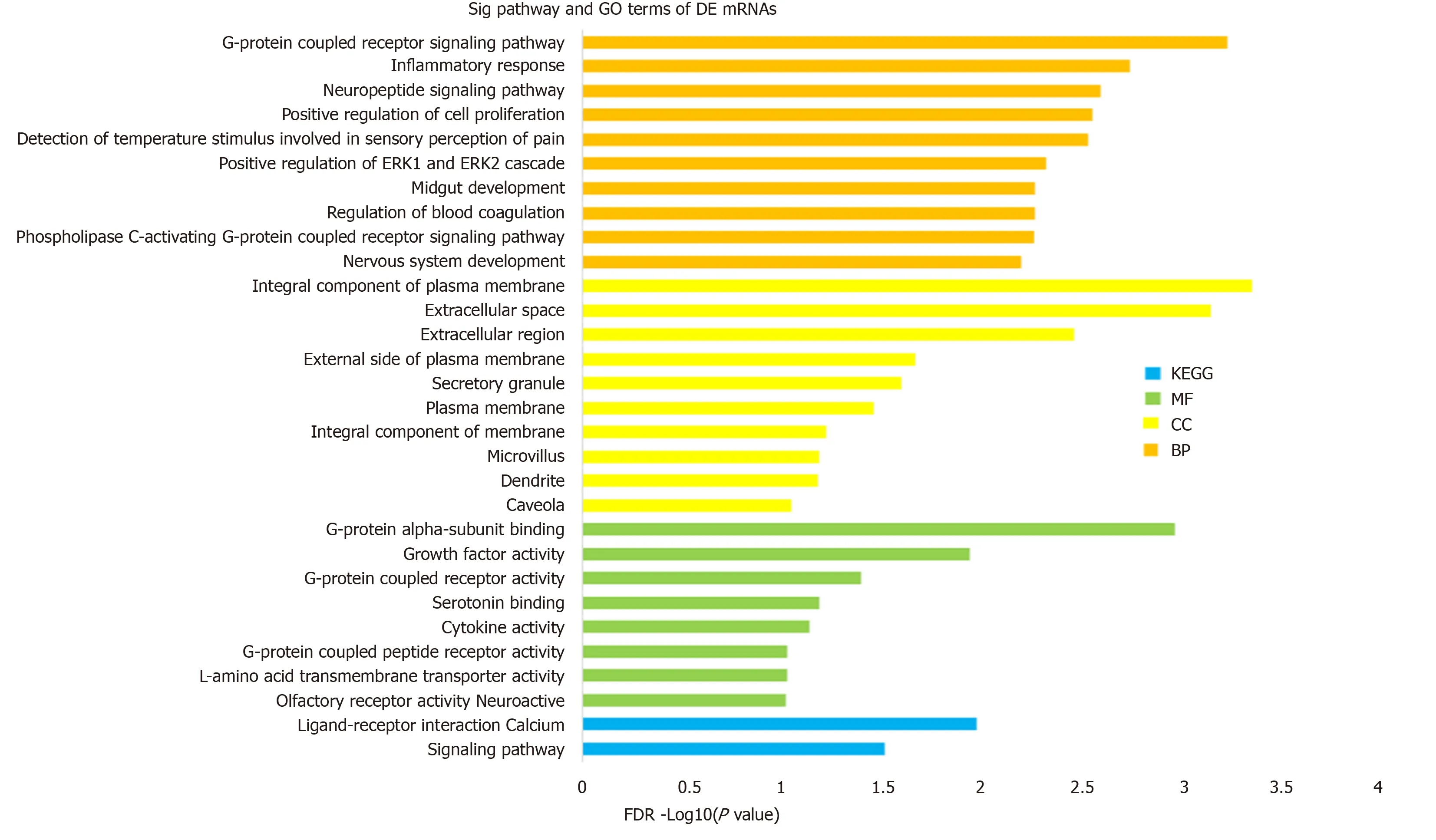
Figure 2 Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment of differentially expressed mRNAs.
Pomc,Htr2a, andAgtr1awere also identified as hub mRNAs in the ceRNA network of pancreatic islet-like cell clusters.Pomcexpression was downregulated in T3pi cells,which could increase β cell proportions and insulin synthesis[28]. Dominguezet al[29]compared the mRNA expression in pancreatic islets from type 2 diabetic and nondiabetic patients. The mRNA expression levels were higher in T2DM patients;however, their findings confirmed that the lower expression ofPomcwas protective to β cells. It was reported that the expression of 5-hydroxytryptamine (5-HT) participates in the regulation of insulin secretion, and overexpression ofHtr2ais associated with islet dysfunction in T2DM[30]. Testosterone was shown to prevent pancreatic β cell apoptosis by suppressingAgtr1aexpression[31]. These studies further confirm our finding that the downregulation ofPomc,Htr2a, andAgtr1ais protective to β cells.
Based on the GO and KEGG analyses,Pomc,Htr2a, andAgtr1awere enriched in the G-protein-coupled receptor signaling pathway, serotonin receptor signaling pathway,inflammatory response, positive regulation of cell proliferation, ERK1 and ERK2 cascade, and cytosolic calcium ion concentration. More studies have shown that the ERK signaling pathway[32], calcium ion concentration[33], inflammatory response[34], and cell proliferation are essential for augmenting insulin secretion and β cell mass protection from premature apoptosis.
Additionally, through the SwissTargetPrediction database, we verified thatHtr2aandAgtr1aare GLP-1 and geniposide targets. This result strongly confirmed the accuracy of our screening for hub mRNAs. Specifically,Pomcscored highest among the hub mRNAs based on our CytoHubba analysis, suggesting thatPomcis a potential GLP-1RA target.
Our analysis showed that six miRNAs were involved in mediating GLP-1RA function within the ceRNA network. In support of our results, four of the six miRNAs(miR-449a,miR-378a,miR-344, andmiR-874) have already been implicated in regulating insulin signaling, improving metabolic dysregulation, and activating insulin synthesis[35-38]. The other two miRNAs (miR-5132-3pandmiR-3075)have been suggested to play a regulatory role in the proliferation and migration of osteoblasts and Schwann cells[39,40]. Hence, we propose thatmiR-5132-3pandmiR-3075may act as new effector molecules in β-cell regulation.
LncRNAs can act as miRNA sponges to regulate mRNA expression and activityviathe ceRNA mechanism[24]. Recently, studies have revealed that lncRNAs are involved in the process of insulin secretion and β cell apoptosis through the ceRNA mechanism[41,42]. In this study, we identified seven lncRNAs (NONRATT002662,NONRATT005090, NONRATT005619, NONRATT019513, NONRATT027738,NONRATT027888, and NONRATT030038) that competitively bind to six miRNAs(rno-miR-449a-5p, rno-miR-5132-3p, rno-miR-344g, rno-miR-3075, rno-miR-378a-5p,and rno-miR-874-3p), thereby influencing the expression and function of hub mRNAs (Pomc,Htr2a, andAgtr1a). Deciphering this lncRNA-miRNA-mRNA network deepens our understanding of the ceRNA mechanism in the protective effect of GLP-1RA in β cells.
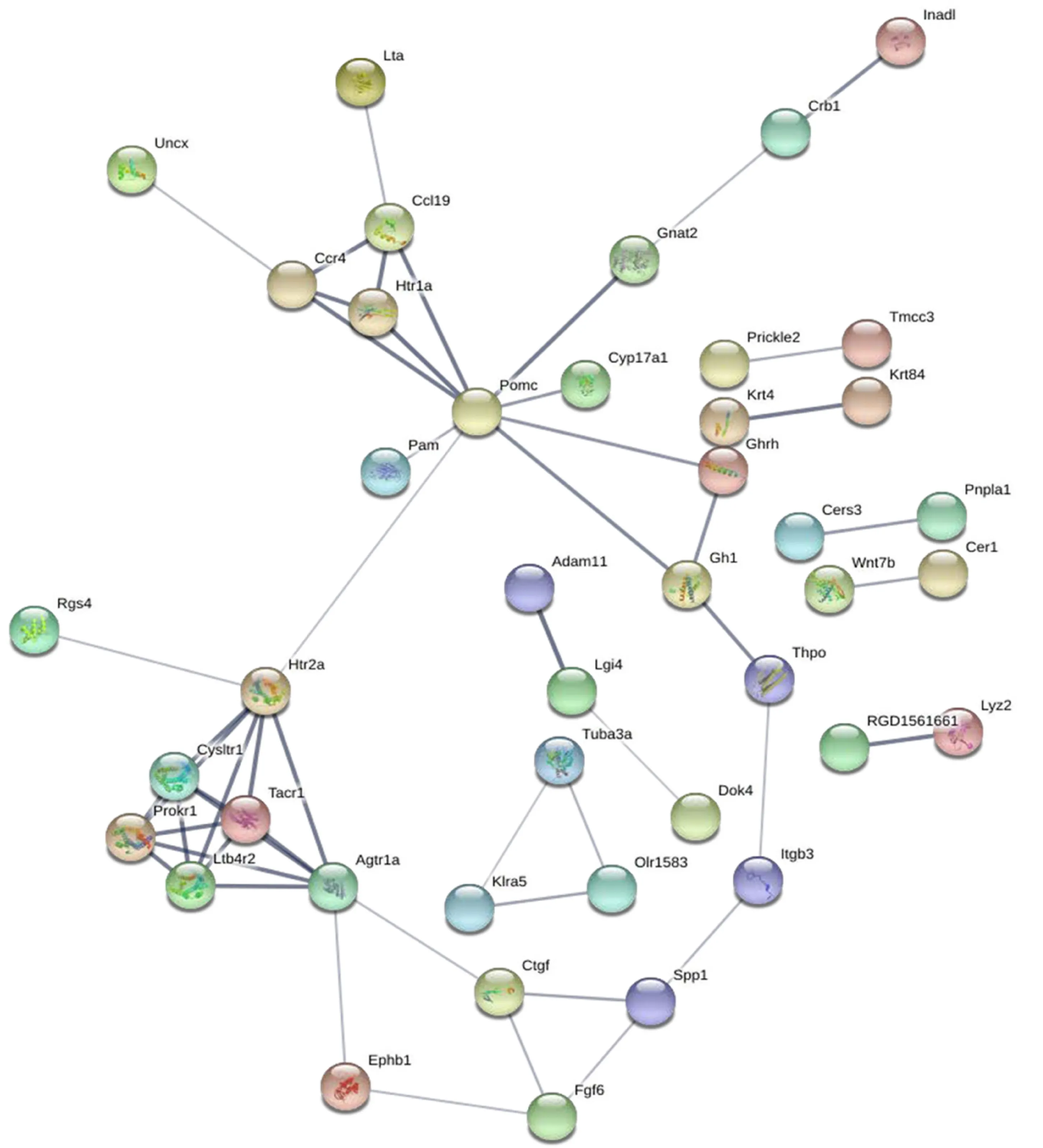
Figure 3 Protein-protein interaction network of differentially expressed mRNAs.
Considering that therapeutic RNAi technology has now been tested in humans[43,44],we believe that our report provides novel RNAs as potential therapeutic targets in the GLP-1RA-mediated protection of β cells. However, further studies are required to better understand and confirm the specific function of these RNAs in β cells.
In summary, this study revealed the expression profiles of lncRNAs and mRNAs in geniposide-treated INS-1 cells. Further explorationviabiological information analysis demonstrated that the ceRNA mechanism is involved in the regulatory relationship between lncRNAs and mRNAs in β cells. These findings provide significant insight in understanding the mechanisms of GLP-1RA function at the transcription level.
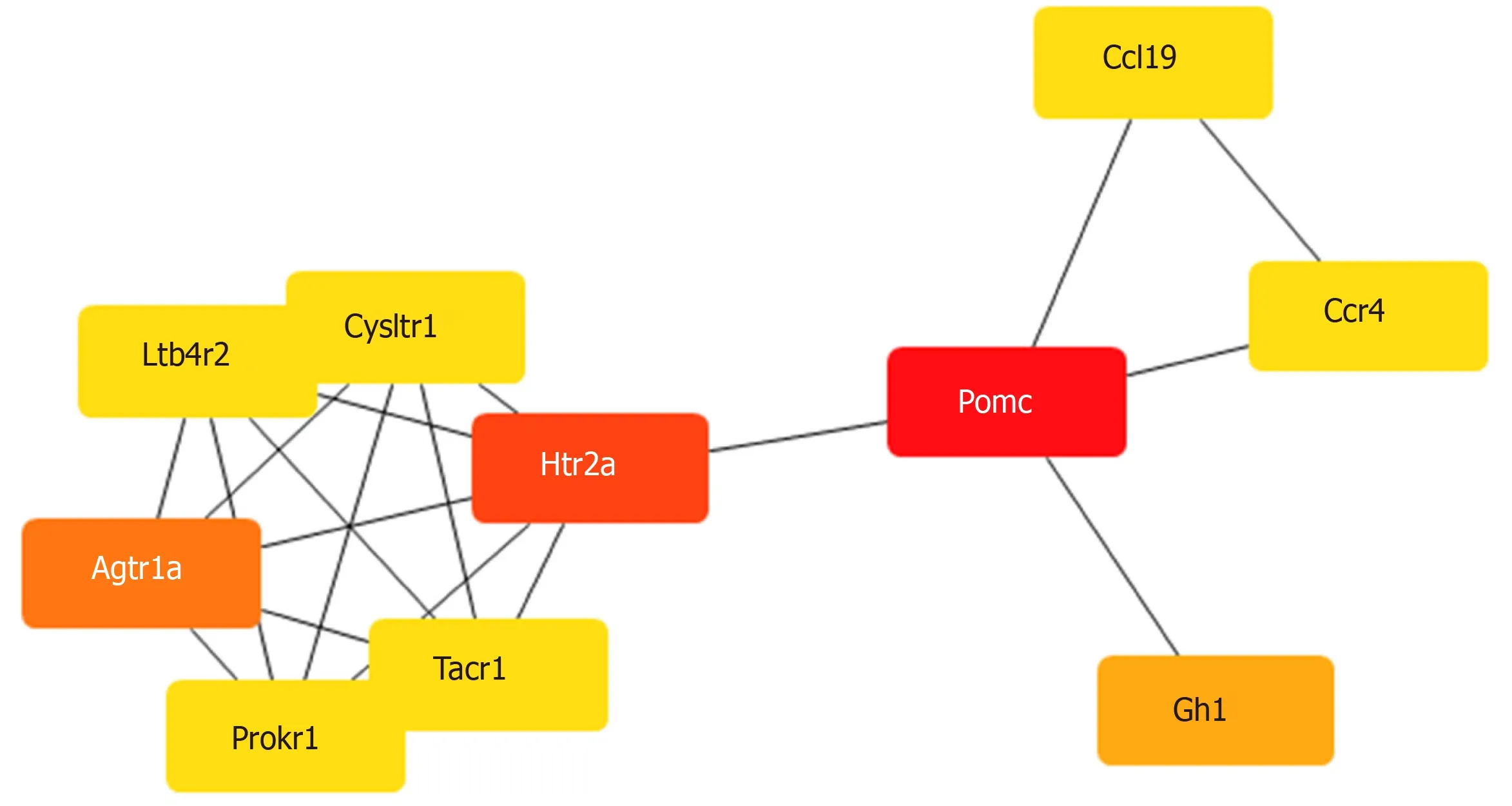
Figure 4 Hub mRNAs.
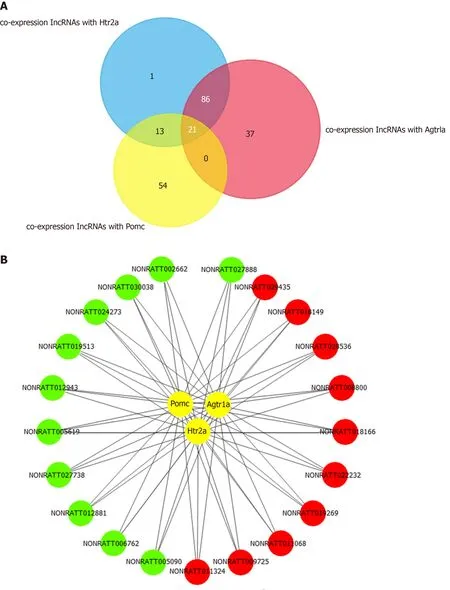
Figure 5 Co-expression network of long noncoding RNAs and hub mRNAs.
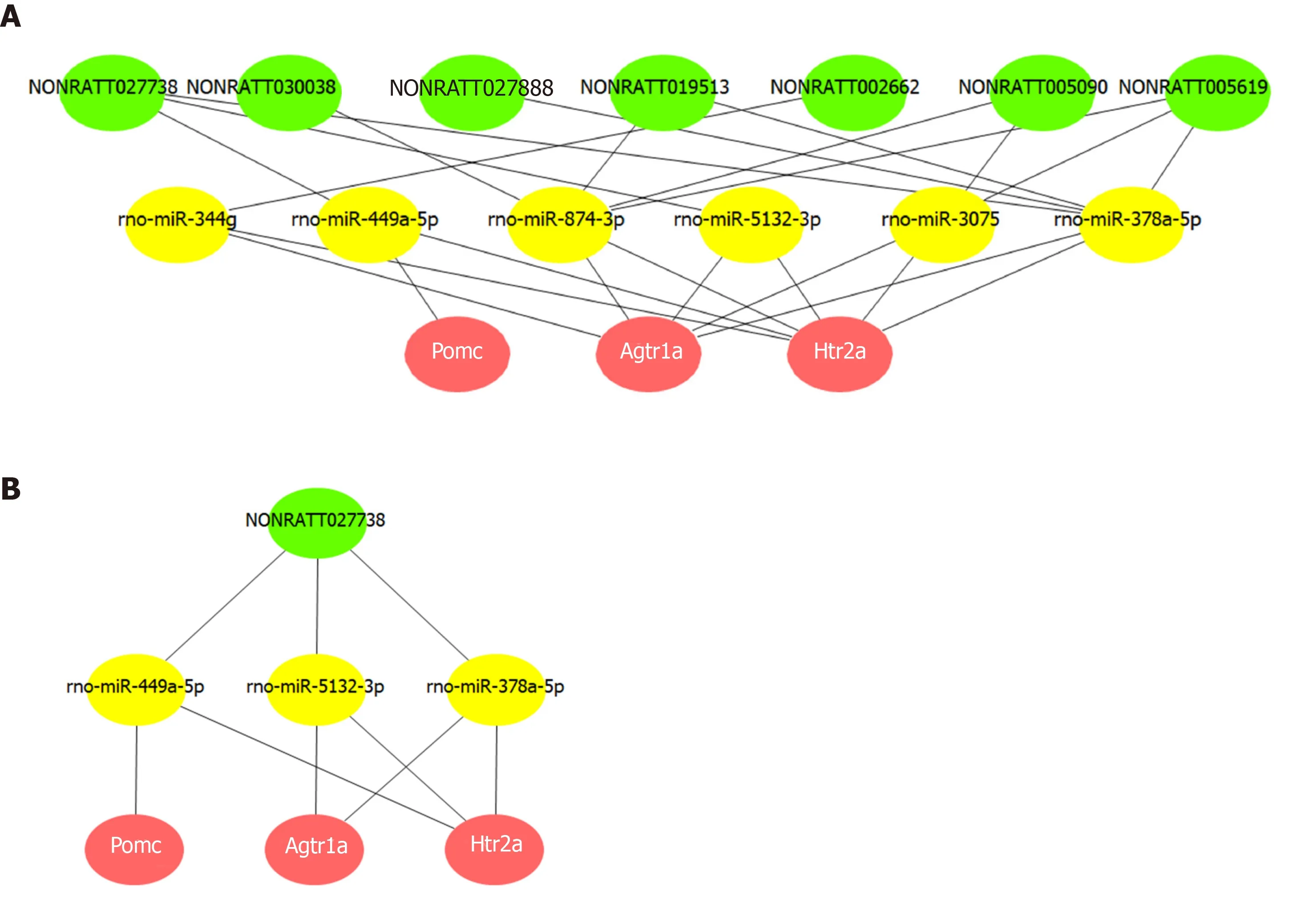
Figure 6 Competing endogenous RNA network of key long noncoding RNAs and Pomc, Htr2a, and Agtr1a.
ARTICLE HIGHLIGHTS
Research background
As a class of promising anti-diabetic drugs, glucagon-like peptide-1 receptor agonists(GLP-1RAs) have been shown to prevent β cells from apoptosis and potentiate insulin secretion in a glucose-dependent manner, which can decrease blood glucose levels without the risk of hypoglycemia. Long noncoding RNAs (lncRNA) are transcripts that are longer than 200 nucleotides and do not code for proteins. Growing evidence demonstrates that lncRNAs regulate mRNA expression by competing with miRNAs,which was termed as “ceRNA mechanism”. Studies have demonstrated that ceRNA mechanism is widely involved in multiple biological processes, including insulin signal transduction that may affect diabetes development. Currently, the mechanisms of the protective effect of GLP-1RA on β cells have been widely investigated; however,the specific lncRNAs and mRNAs and their functions in these processes have not been fully identified and elucidated.
Research motivation
Is there any specific lncRNAs that participate in the protective effect of GLP-1RAs in β cells? What is the mechanism of lncRNAs involved in this process? Answering these questions will provide significant insight in understanding the mechanisms of GLP-1RA function at the transcription level.
Research objectives
We and other researchers found that geniposide potentiates insulin secretion,promotes proliferation, and decreases the rate of β cell apoptosis by stimulating the GLP-1 receptor. In this study, we further identified the lncRNAs and mRNAs that were involved in the protective effect of geniposide on β cells, and their roles. This will be helpful for in-depth exploration of the mechanism of GLP-1RAs function in β cells.
Research methods
Rat gene microarray was used to screen differentially expressed (DE) lncRNAs and mRNAs in β cells treated with geniposide, a GLP-1RA. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to assess the underlying functions of DE mRNAs. Hub mRNAs were filtered using the STRING database and the Cytoscape plugin, CytoHubba. In order to reveal the regulatory relationship between lncRNAs and hub mRNAs, their coexpression network was constructed based on the Pearson coefficient of DE lncRNAs and mRNAs, and competing endogenous (ceRNA) mechanism was explored through miRanda and TargetScan databases.
Research results
We identified 308 DE lncRNAs and 128 DE mRNAs with a fold change filter of ≥ 1.5 andPvalue < 0.05. GO and KEGG pathway enrichment analyses indicated that the most enriched terms were G-protein coupled receptor signaling pathway,inflammatory response, calcium signaling pathway, positive regulation of cell proliferation, and ERK1 and ERK2 cascade.Pomc,Htr2a, andAgtr1awere screened as hub mRNAs using the STRING database and the Cytoscape plugin, CytoHubba. This result was further verified using SwissTargetPrediction tool. Through the coexpression network and competing endogenous (ceRNA) mechanism, we identified seven lncRNAs (NONRATT027738, NONRATT027888, NONRATT030038,etc.) coexpressed with the three hub mRNAs (Pomc, Htr2a, and Agtr1a) based on the Pearson coefficient of the expression levels. These lncRNAs regulated hub mRNA functions by competing with six miRNAs (rno-miR-5132-3p, rno-miR-344g, rno-miR-3075,etc.)viathe ceRNA mechanism. Further analysis indicated that lncRNANONRATT027738interacts with all the three hub mRNAs, suggesting that it is at a core position within the ceRNA network.
Research conclusions
We have identified key lncRNAs and mRNAs, and highlighted here how they interact through the ceRNA mechanism to mediate the protective effect of GLP-1RA in β cells.
Research perspectives
The “ceRNA mechanism”, which is widely involved in multiple biological processes,mediates the protective effect of GLP-1RAs in β cells. The value of bioinformatics allows scientists to create comprehensive databases of biological and health information that can be used to test theories and generate solutions to medical problems that affect us all.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Tao Bai for skillful technical assistance.
