Review: Pathogenesis of cholestatic liver diseases
2021-01-14RaquelYokodaEduardoRodriguez
Raquel T Yokoda, Eduardo A Rodriguez
Raquel T Yokoda, Department of Anatomic and Clinical Pathology, Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY 10467, United States
Eduardo A Rodriguez, Department of Gastroenterology, Hepatology and Nutrition, University of Utah, Salt Lake City, UT 84132, United States
Abstract Cholestatic liver diseases (CLD) begin to develop after an impairment of bile flow start to affect the biliary tree. Cholangiocytes actively participate in the liver response to injury and repair and the intensity of this reaction is a determinant factor for the development of CLD. Progressive cholangiopathies may ultimately lead to end-stage liver disease requiring at the end orthotopic liver transplantation. This narrative review will discuss cholangiocyte biology and pathogenesis mechanisms involved in four intrahepatic CLD: Primary biliary cholangitis, primary sclerosing cholangitis, cystic fibrosis involving the liver, and polycystic liver disease.
Key words: Cholestasis; Cholangitis; Epigenomics; Immunogenetics; Pathogenesis; Bile acid
INTRODUCTION
Cholestatic liver diseases (CLD) encompasses progressive cholangiopathies, which may evolve to end-stage liver disease. In the United States from 1988 to 2018, this group of illness corresponded to 14.2% of all liver transplants[1]. Thus far, their high morbidity and mortality are an economic burden that evolved from the lack of effective treatments. Moreover, 10% to 40% of these patients will have a recurrence of the primary disease after liver transplantation (LT)[2].
New prospective therapeutic targets are an unmet necessity, a number of which are under preclinical development. To evaluate these potential therapies, it is essential to understand the primary target of these pathologies, the cholangiocytes. This review will reinforce the current understanding of the core concepts of CLD pathogenesis in the light of the last translational advancements that may impact clinical management.
CLD: COMMON PATHOGENIC MECHANISMS
Several factors can condition bile flow derangements (Figure 1). Although environmental triggering factors are mostly unknown, antigenic stimuli, exotoxins, endotoxins, xenobiotics, and microorganisms can promote cholangiocyte reaction that will evolve into a cholestatic state[3]. Bile transport obstruction is another predisposing factor. Intrahepatic and extrahepatic obstruction can take place due to extrinsic benign compression (cystic diseases), malignant mass effect (cholangiocarcinomas), and also as a consequence of cholelithiasis formation or migration throughout the biliary tree. Moreover, conditions that slow biliary flow promote a cholestatic state with increased bile acid (BA) concentration. Sepsis, hyperestrogenic states (pregnancy), congestive heart failure, and dysfunction of BA transporter genes may alter the main characteristics of BA, conditioning a more cytotoxic BA component.
Early cholangiocyte response may allow resolution of injury, however, sustained pro-inflammatory signaling associated with disragulation of genetic and/or epigenetic regulatory mechanisms could condition late dysfunctional permanent state. Eventually fibrogenic state with biliary and periportal fibrosis, loss of tissue homeostasis and autocrine and paracrine remodeling would be achieved. Ultimately, proliferation may lead to cell-cycle alteration, senescence, apoptosis, ductopenia, mesenchymal infiltration and sometimes malignant transformation. To date, new therapeutic targets are being developed for each CLD considering the core of this pathogenic process. The main framework will be analyzed along with the foundation for potential clinical development.
Ductular reaction: First core concept
Intra and extra-hepatic bile ductules of different sizes are lined by cholangiocytes, which are epithelial cells that regulate and modify bile volume and composition[3]. These vary in size, metabolic rate as well as proliferative and plasticity capabilities. Biliary differentiation pathways are being more thoroughly understood and so it is now known that hepatocytes and cholangiocytes have a common stem cell precursor, and trans differentiation may occur in massive parenchymal loss from one to another, although the exact mechanisms are not well understood[4].
Ductular reaction (DR) is part of the injury response. It is triggered by cholestasis which activates the hepatic progenitor cells in CLD[5]. The sonic-hedgehog pathway promotes both cholangiocyte maturation and deposition of fibronectin in ductularreactive cells[6]. DR may induce injury resolution, or, biliary fibrosis in the presence of perpetuating transcriptional inflammatory addiction. The cytokine panel for this transcriptional impairment depends on the disease phenotype and ultimately will condition different histological classifications beyond the scope of this review[7]. Figure 2 lists the dominant spectrum of CLD.
Bile acid toxicity and mitochondrial dysfunction
The second core fundamental framework of CLD pathogenesis is BA cytotoxicity and mitochondrial dysfunction. Besides its functional role of converting lipid bilayers into mixed micelles, BA are endogenous ligands that activate a network of receptors including nuclear receptor farnesoid X (FXR), vitamin D3 receptor (VDR), pregnane X receptor (PXR), constitutive androstane receptor (CAR), membrane G protein-coupled bile acid receptor-1, and Takeda-G-protein receptor5 (TGR5). Indeed, FXR and TGR5 provide an anti-inflammatory liver response in mouse models[8]. In fact, FXR mutations have been considered a cause of progressive familial intrahepatic cholestasis. Intestinal activation of FXR increases FGF15, a bile synthesis repressor through CYP7A1, a main regulatory enzyme, which reduces the pool size of BA and protects against escalating pro-inflammatory signaling in mouse models[9].
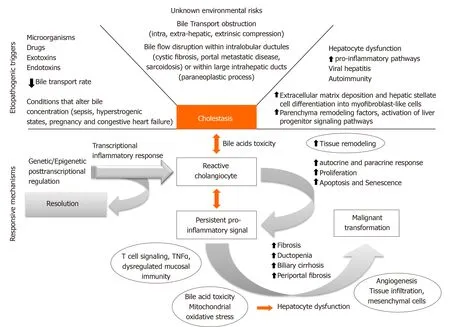
Figure 1 Core pathogenic mechanism of cholestatic liver diseases.
Likewise, BA hepatobiliary transport dysfunction may lead to several phenotypes of cholestatic diseases. Although transcellular BA transport details are mostly unknown, a number of apical and basolateral transporters have been identified. After synthesis of BA in the liver by CYP7A1 and hydroxylation by CYP8B1, bile acids and phospholipids are excreted and secreted across the canalicular membrane of hepatocytes into the biliary tree by BSEP (bile salt export pump/ABCB11) and ABCB4 (ATP binding cassette subfamily B member 4), respectively. BA are then re-uptaken in the terminal ileum by ASBT (apical sodium-dependent bile acid transporter/ SCL10A2), and released into the portal system by a basolateral transporter (OSTα/β) and may later be re-uptaken by the liverviaNTCP (Na+/taurocholate cotransporting polypeptide) or OATP (organic anion transporting polypeptides) transporters. Intrahepatic BA can further be processed by hydroxylation, glucuronidation or sulfation, and excreted back into sinusoidal and systemic circulation by OSTα/β and MRP3/4 bile acid transporters. Critical steps in the enterohepatic circulation are regulated by the BA receptor FXR, which limits BA uptake and synthesis by enhancing biliary and basolateral BA export. FGF19, a gut-derived FXR-dependent enterocrine hormone, suppresses hepatic bile acid synthesis and induces gallbladder filling when it is activated by high intestinal BA concentrations[10].
Recently, AMP-activated protein kinase (AMPK) signaling pathways have been implicated in the pathogenesis of drug-induced cholestasis[11]. An example of this pathway is metformin. An older study reported that after 2-3 wk of metformin usage, several patients developed portal inflammation and ductular proliferation[12].
Moreover, it is well-known that the hydrophilic profiles in BA spectrum protects against apoptosis (TCA and UDCA), while those in the hydrophobic range induce hepatic apoptosis and liver injury (TLCA and GCDCA). Additionally, accumulation of cytotoxic BA activates NF-κB-mediated inflammatory cytokines. This pathway is significant in intrahepatic cholestasis of pregnancy as it may arrest placental inflammation[13].
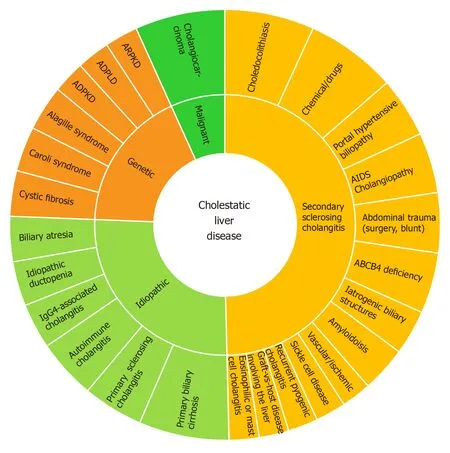
Figure 2 Cholestatic liver disease clinical spectrum.
Several studies have described BA toxicities and established commonalities between this toxicity and mitochondrial dysfunction in extra-hepatic cholestasis[14].In vitrostudies demonstrated BA effect in normal liver cell line LO2. Glycochenodeoxycholic acid (GCDCA) stimulated cytotoxicity, disrupted the mitochondrial membrane potential, increasing production of reactive oxygen species (ROS), and leading to decreased mitochondrial mass and mitochondrial DNA content[14]. This feature can be fundamentally related to the development of anti-mitochondrial antibodies (AMA) in primary biliary cholangitis (PBC), consequence of infiltration by both CD4+ and CD8+ T cells reactive to conserved mitochondrial and nuclear antigens, particularly the E2 component of the pyruvate dehydrogenase complex — the principal target of circulating AMA[15]. Moreover, one study pointed deacetylation of the gene PGC-1α, peroxisome proliferator-activated receptor gamma, coactivator one alpha. PGC-1 α acts as an enzyme in mitochondria biogenesis[14]. In chronic intrahepatic cholestasis, the lipid peroxidation activates extracellular matrix cells, ROS, and aldehydes; which may exert direct fibrogenic effects on activated hepatic stellate cells[16].
Immunogenetic and epigenetic setpoints
The third fundamental aspect of the core framework is the influence of immunogenetics and epigenetics on immunoinflammatory response. Patients with CLD exhibit a variety of genetic alterations that account for the different elements of each CLD. However, some of those genes may be directly implicated in the progression rate of the cholestatic phenotype. Recently one study screened some of the progression-related candidate genes for primary biliary cholangitis[17]. They evaluated 315 DNA samples from patients for single nucleotide polymorphisms (SNPs) of 11 candidate genes involved in regulation of bile acid synthesis. Interestingly, genetic variants of CYP7A1, as well as its transcriptional activators (HNF4A and PPARGC1A), may activate bile acid synthesis in an escalating fashion leading to the progressing cholestasis in PBC[17]. It is significant that this gene could become a potential target for new therapeutics, or indirectly their transcriptional activators could serve as modulatory targets. This modulation is a type of epigenetic control of gene expression as a pathogenic mechanism.
Another study highlighted the central role of the IL-12-STAT4-Th1 pathway, a proinflammatory pathway in the progression of PBC, as well as the HLA associations and epigenetic effects[18,19]. Figure 3 shows a panel of immunogenetic genes, where those directly related to the T-cell function or the B-cells or the IL12-STAT4-Th1 are highlighted with a red dot. Additionally, genes associated with loss of immunetolerance and epithelial permeability are marked with a yellow dot[20,21].

Figure 3 Immunogenetics related to the core of cholestatic liver diseases.
Dysfunctional matrix re-arrangements and fibrogenesis
To complete the core framework of CLD, dysfunctional matrix rearrangements and fibrogenesis are the fourth concept. Fibrogenesis is a dynamic process that appears intricate to immunoinflammatory mechanisms, secretion of tissue metalloproteinases, cytokine networks and derangements of mesenchymal cells infiltration with ultimate loss of tissue maintenance homeostasis[16]. The pattern of extra cellular matrix (ECM) accumulation in some CLD such as PBC is characterized by increased expression of mRNA encoding collagen type I, III, and IV, which in mesenchymal cells promotes the expansion of portal tracts, leading to deposition of excessive fibrillar ECM. In this way the fibrogenic processes involve damaged and non-damaged bile ducts as well as the periportal sinusoidal system, resulting in progressive cholestasis[16]. In contrast in patients with primary sclerosing cholangitis (PSC), the fibrogenic process has been compared to atherosclerosis onion-like concentric recruitment of pro-fibrogenic cells. Also animal models have reported vascular injury with ischemia of the bile duct epithelial cells during development of PSC lesion[22].
Hepatic stellate cells (HSC) are the primary source of myofibroblast during liver injury, however mesenchymal cells also give rise to myofibroblasts (portal myofibroblasts (PMF) as these cells are located in the portal tract)[23]. Studies in animal models of biliary cirrhosis (rat) reported that PMF use vascular endothelial growth factor A-containing microparticles signaling for newly formed vessels, driving scar progression, while acting as mural cells[24]. This type of fibrosis progression originating from the portal tract is crucial in cystic fibrosis-related liver fibrosis[25]. In PBC epigenetic influence has been observed in the discordance of monozygotic twins. The role of the CD40-CD40L interaction in T-cell and B-cell mechanisms has been reported in the decreased methylation of CD40L promoter regions amongst PBC patients compared with controls[18]. Similarly, X chromosome monosomy has been found on peripheral cells of PBC patients[26]. Recently the Milan PBC epigenetic Study Group reported demethylation of the CXCR3 promoter, which is negatively correlated with peripheral blood receptor expression in CD4+ T-cells[27]. The epigenetic role of demethylation is considered as CXCL9-11 is up-regulated in damaged bile ducts and it is a co-ligand for CXCR3, which is highly expressed in Th1 and Th17[28]. Another group evaluated the role of microRNA (miR), that can also promote downregulation of protein-coding gene expression. Down-Regulation of miR-122a and miR-26a was reported, as well as an increased expression of miR-328 and miR-299-5p. These microRNAs are known to affect cell proliferation, inflammation, oxidative stress metabolism, and apoptosis[29].
PRE-CLINICAL THERAPEUTIC DEVELOPMENTS
From a pathogenic standpoint, a number of therapeutic genetic and epigenetic targets can be considered. Some pathways already have one or more target drugs available. Table 1.
A number of preclinical studies may pave the way to new clinical advancements. A few of them are listed in Table 2, where we highlight the main pathogenic framework as described before.
CLINICAL TRIALS AND TRANSLATIONAL RESEARCH
The core fundamental concepts and pathogenic framework are platforms to build new models of clinical interventions for specific CLD. This section addresses the main cholestatic diseases individually.
Primary biliary cholangitis
PBC is characterized histologically by intralobular nonsuppurative bile duct destruction by lymphocytic cholangitis[30]. Patients with PBC often have a decreased quality of life as the disease progresses to hepatic fibrosis and end-stage liver disease. To date, one-third of the patients do not have a biochemical response to ursodeoxycholic acid (UDCA), which is primarily defined by bilirubin and alkaline phosphatase levels after one year of UDCA.
PBC inflammatory disarrays present with increased cholangiocyte chemokines released mainly CXCL10, CXCL9, CX3CL1, and CCL20, which involve the IL-12/IL23 pathways[31]. A number of novel therapeutics in immunomodulation such as fibrates and budesonide had promising results as an alternative to UDCA nonresponders, and recently obeticholic acid was approved by the FDA for UDCA non responders[32-34]. Advancements for PBC patients also include agonists for peroxisome proliferatoractivated receptor alpha (PPARα), FXR, GR/PXR most often in combination with UDCA, fibrates, obeticholic acid (OCA) and budesonide, respectively[35]. Some of these translational therapeutics are mentioned in Table 3 and can also be used in PSC as discussed as follows.
Primary sclerosing cholangitis
There are currently no approved therapies for PSC. The disease causes a significant economic burden, and patients have high hospitalization and malignancy rates, often progressing to end-stage liver disease, requiring eventually liver transplantation. Table 3 summarizes the main translational research in the field. Novel approaches for PSC include transcriptional modifiers of bile formation, such as the agonists of FXR, PXR, GR and activation of PPARα. This activation can be promoted by fibrates as they decrease expression of inflammatory cytokines, also reducing hepatocyte BA synthesis. Another approach is the use of agonists of Takeda-G-protein 5 (TGR5), a BA membrane receptor expressed in various tissues as it can lower the levels of proinflammatory cytokines in bile ducts[36]. Other approaches include inhibitors of the ileal apical sodium BA transporter, derivatives of the FXR-induced fibroblast growth factor 19 (FXR-induced FGF19) from the ileum that suppress hepatic BA synthesis, and norursodesoxicholic acid (norUDCA), a side chain shortened UDCA derivative.
Cystic fibrosis involving the liver – hepatobiliary spectrum
The frequency of biliary manifestations in cystic fibrosis (CF) is still unclear. Clinical phenotypes range from gallbladder dyskinesia, symptomatic cholelithiasis to sclerosing obstructive cholangitis. Early diagnosis can be challanging. Tools like the Aspartate Aminotransferase-to-Platelet Ratio Index (APRI) are reliable at predicting severe fibrosis, but not for differentiating fibrosis in early stages. Therefore, serum biomarkers are an unmet necessity thus far. Promising research areas include further investigating the role of intestinal bile salt malabsorption such as the plasma fibroblast growth factor 19 (FGF19) and the intermediate of CYP7A activity and the 4-cholesten-3-one (C4)[37]. Transient elastography may be useful as well, however appropriate validation in mild-to-moderate fibrosis is still pending[38]. Clinical trials for CF cholestasis, using the new generation of therapeutic targets beyond UDCA, would also provide benefits to patients. Some agents discussed previously had good results in preclinical research, such as NorUDCA, tested in mice[39].
Recent CF animal model investigations uncovered the underpinning relationship of the CF transmembrane conductance regulator and the control of biliary epithelialinflammation and permeability mediated by TLR4-NF-κB[40]. Moreover, a number of studies have identified a dysfunctional PPAR-gamma (peroxisome proliferatoractivated receptor gamma), that was partially recovered with PPAR-gamma ligands, as rosiglitazone, particularly attenuating biliary fibrosis in CF[41]. Another study, also in murine model, linked those PPAR-gamma as a limiting factor for NF-κB-dependent inflammation[42]. These findings can possibly be further studied as possible target for future therapies.

Table 1 Potential pathways as targets for existing antibodies
Polycystic liver disease
Polycystic liver diseases are autosomal dominant disorders that result from a mutation of PRKCSH or Sec63 genes; genes that are mainly expressed in cholangiocytes[43]. Cystogenesis in this scenario is due to benign cholangiocyte proliferation, with cellcycle dysregulation and increased level of cAMP in cholangiocytes leading to cyst progression and abnormal fluid transport[44]. Over time, the cyst growth may compress the biliary tree impairing bile flow as well. Liver volume is a prognostic marker as complications may occur as the disease progresses, such as hepatic cyst infection, rupture, hemorrhage and hepatic venous outflow obstruction[45]. Therapeutic developments have focused in preclinical studies in lowering cAMP and stopping or reversing progression, usually evaluated by the organ size and hepatic cystic volume. Octreotide became an option for treatmentviadecrease in cAMP levels[46,47]. Recently an open-label clinical trial tested UDCA effect in cystic liver diseases and reported a reduction of liver cyst volume growth after 24 wk of treatment[48,49]. This effect was expected as UDCA decreases the concentration of cytotoxic BA and therefore diminishes proliferation stimuli[50]. Additionally, more than 50% of patients may have fibrosis[51].
CONCLUSION
Although CLD pathogenic features are becoming unveiled, and translational research is achieving success, some findings still challenge what we know about the basic molecular developments in CLD, such as the relationship of FXR agonists, synthesis of FGF19 and metabolism expression and cell survival[52], and ultimately possible carcinogenesis. To date, inhibitors of the FGF19/FGFR4 pathway are in development for the treatment of hepatocellular malignancies. This acknowledgment for the regular hepatology practice is essential, as for a number of cases, hepatologists and oncologist specialized in hepatobiliary tumors do not often work on the same cases at the same point in time. However, the same patient may experience interactions with these professionals on different occasions in the course of disease progression. For the current therapeutics of cholestatic disease, FXR agonists may represent a novel approach for PBC, and trigger experimentational use for PSC. In the long run, however, the aberrant expression of FGF19 in its oncogenic driver is not entirely presumed. The landscape of modulation of the fibroblast growth factor family, as wellas its signal through the transmembrane tyrosine kinase receptors, needs an operable spotlight in cholestatic diseases.

Table 2 Preclinical research cholestatic liver diseases
Moreover, in pre-carcinogenic sclerosing conditions such as PSC, the agonistic effect of cell proliferation, differentiation, and tissue repair through a potential oncogenic signaling pathway demands further scrutiny. Besides, a possible role in therapeutic resistance for advanced metastatic hepatocellular carcinomas, once the pathway is wired up, is also concerning. Epigenetic modulation in the core of the CLD and the hepatostat growth activation through FGF19/FGFR4 may interface with the Hippo-Yap signaling and play an essential role in liver carcinogenesis.
It is expected that the current understanding of the multifactorial pathogenic process and the potential substantial role of epigenetics will drive further much needed basic research and introduce new concepts and prospective therapeutic targets to the world of CLD.

Table 3 Clinical trials and translational research
杂志排行

World Journal of Hepatology的其它文章
- Mechanisms and consequences of COVID-19 associated liver injury:What can we affirm?
- Lipidomics in non-alcoholic fatty liver disease
- Update on diagnosis and management of sepsis in cirrhosis: Current advances
- Cell competition in liver carcinogenesis
- Management of hepatitis C in children and adolescents during COVID-19 pandemic
- Glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: An update