Characteristics of viral specimens collected from asymptomatic and fatal cases of COVID-19
2021-01-13AndrewGorzalskiPaulHartleyChrisLaverdureHeatherKerwinRichardTillettSubhashVermaCyprianRossettoSergeyMorzunovStephanieVanHooserMarkPandori
Andrew J. Gorzalski, Paul Hartley, Chris Laverdure, Heather Kerwin, Richard Tillett,Subhash Verma, Cyprian Rossetto, Sergey Morzunov, Stephanie Van Hooser, Mark W. Pandori,7,✉
1Nevada State Public Health Laboratory, Reno, NV 89597, USA;2Nevada Genomics Center, Reno, NV 89557, USA;3Washoe County Health District, Reno, NV 89512, USA;4Nevada Institute of Personalized Medicine, University of Nevada Las Vegas, Las Vegas, NV 89154-4022, USA;5Nevada Center for Bioinformatics, University of Nevada Reno, Reno, NV 89557, USA;6Department of Microbiology and Immunology, 7Department of Pathology and Laboratory Medicine, University of Nevada, Reno School of Medicine, Reno, NV 89557, USA.
Abstract We sought to determine the characteristics of viral specimens associated with fatal cases, asymptomatic cases and non-fatal symptomatic cases of COVID-19. This included the analysis of 1264 specimens found reactive for at least two SARS-CoV-2 specific loci from people screened for infection in Northern Nevada in March-May of 2020. Of these, 30 were specimens from fatal cases, while 23 were from positive, asymptomatic cases. We assessed the relative amounts of SARS-CoV-2 RNA from sample swabs by real-time PCR and use of the threshold crossing value (Ct). Moreover, we compared the amount of human RNase P found on the same swabs.A considerably higher viral load was found to be associated with swabs from cases involving fatality and the difference was found to be strongly statistically significant. Noting this difference, we sought to assess whether any genetic correlation could be found in association with virus from fatal cases using whole genome sequencing.While no common genetic elements were discerned, one branch of epidemiologically linked fatal cases did have two point mutations, which no other of 156 sequenced cases from northern Nevada had. The mutations caused amino acid changes in the 3′-5′ exonuclease protein, and the product of the gene, orf8.
Keywords: COVID-19, SARS-COV-2, real-time PCR, Ct value, viral load
Introduction
Laboratory diagnostics utilized through the early phases of the COVID-19 pandemic have relied entirely on the use of real-time PCR for direct detection of SARS-CoV-2. While other molecular and protein-based methodologies are becoming available,real-time PCR will likely remain the dominant mechanism of detection due to widespread availability of relevant equipment in diagnostic labs, and the ease by which such methods can be instituted in laboratories. While real-time PCR provides a sensitive means of detection in a Boolean manner(presence/absence), most real-time PCR systems provide a quantitative assessment of a specimen in the form of "threshold cycles" often referred to as Ct values. This value is inversely proportional to the amount of target detected in a real-time PCR reaction and refers to the intersection between an amplification curve and a designated threshold of fluorescence associated with background[1]. In certain real-time diagnostic usages, such as with the detection of human immunodeficiency virus or hepatitis viruses (B and C), these Ct values are correlated to values on a generated standard curve, allowing viral loads to be ascertained on the basis of the number of genomes detected. Execution of this task is relevant to the medical management of these diseases, because therapeutic treatment requires detailed monitoring to assess adherence to pharmacological therapy and the development of resistance. For virtually all other usages, attention to Ct values is generally not considered for lack of any correlative medical assessment.
COVID-19 disease is associated with a wide range of outcomes, ranging from lack of any apparent illness(asymptomatic disease) to death. The majority of cases manifest with some combination of fever,cough, and fatigue, but other symptoms including that loss of taste and smell, sore throat and enteric illness were also described. Fatal cases of COVID-19 have been shown to be associated with a variety of host factors, including hypertension, coronary heart disease, diabetes and age among other factors[2]. It is currently unknown whether disease severity is linked to viral factors, such as infective dose or viral genetics. Certain genotypes have been associated with increased success, including enhanced transmissibility[3–5]. To date, however, with no certainty have certain genotypes been associated with more severe medical outcomes however a singular mutation may be so associated[6]. Viral load on test collection swabs (as estimated by Ct value) has been associated with increased severity of lung disease in COVID-19[7].Moreover, there is evidence for higher loads to correlate with more severe disease outcomes[8–9].We sought to compare the Ct values from real-time PCR to cases with varying outcomes. Noting a difference in the mean Ct values between fatal cases and non-fatal cases, we further assessed the viral genomes associated with such cases, and compared them to the genomes of non-fatal cases. The data of this study are provided herein.
Materials and methods
Real-time PCR
Specimens were collected throughout the state of Nevada (March 1 – May 31, 2020) and included symptomatic individuals (self-reported presence of fever, cough or shortness of breath) or individuals associated with an outbreak at a facility, regardless of symptomology. Specimens were taken by nasopharyngeal swab and transported to the Nevada State Public Health Laboratory in viral transport medium (VTM). Specimens were transported on cold packs and stored by refrigeration (4 to 8 °C) for 72 hours or less prior to being subject to nucleic acid extraction and subsequent real-time PCR. Extraction was performed by Omega Biotek MagBind Viral DNA/RNA 96 Kit following manufacturer's instructions with an elution volume of 100 μL. Eluted RNA (5 μL) was subjected to real-time PCR either by the CDC EUA Real-Time PCR for SARS-CoV-2.This PCR detects two SARS-CoV-2 specific targets deemed, "N1" and "N2".
Viral genomic sequencing
Total RNA was extracted from nasopharyngeal swabs with commercially available kits (QIAGEN,Omega BioTek) designed for the recovery of low abundance RNA. This extracted RNA (30 to 80 μL)was treated for 30 minutes at room temperature with QIAGEN DNase I and then cleaned and concentrated with silica spin columns (QIAGEN RNeasy MinElute), with a 12-μL water elution. A portion(7 μL) of this RNA was annealed to an rRNA inhibitor(QIAGEN FastSelect -rRNA HMR), and then reverse transcribed, strand-ligated and isothermally amplified into micrograms of DNA (QIAGEN FX Single Cell RNA Library Kit). A portion (1 μg of this amplified DNA was sheared and ligated to Illumina-compatible sequencing adapters, followed by 6 cycles of PCR amplification (KAPA HiFi HotStart) to enrich for library molecules with adapters at both ends. Next,these sequencing libraries were enriched for sequence specific to SARS-CoV-2 using biotinylated oligonucleotides (myBaits Expert Virus, Arbor Biosciences). A further 8 to 16 cycles of PCR were performed post-enrichment, and these SARS-CoV-2 enriched sequencing libraries were pooled and sequenced with an Illumina NextSeq 500 as pairedend 2×75 bp reads.
Phylogenetic analysis
Library quality metrics for samples were calculated using FastQC, version 0.11.8[10]. Sequence pairs were trimmed using Trimmomatic, version 0.39, with the ILLUMINACLIP adapter-clipping setting"2:30:10:2:keepBothReads"[11].
Sequence pairs were aligned against the Wuhan reference genome (NC_045512.2) using Bowtie 2,version 2.3.5, in local alignment mode[12]. Alignments were sorted by coordinate using samtools, version 1.9[13]. PCR optical duplicates were removed using Picard MarkDuplicates, picard-slim version 2.22.5[14].Read group tags were added to each sample usingbamaddrg[15]. Quality control, trimming, alignment and deduplication metrics were summarized using MultiQC, version 1.7[16].
Tagged, de-duplicated alignments for every sample were used together to call variants using Freebayes,version 1.3.2, with ploidy set to 1, minimum allele frequency 0.75, and minimum depth of 4[17]. Called variants in in the first 200 bp and final 63 bp of the COVID-19 genome were removed. High-quality variant sites were selected where site "QUAL>1"usingvcffilter, VCFlib version 1.0.0_rc2[18].
Whole genome coverage maps of all samples were reconstructed using bbtools, version 38.86,pileupandapplyvariantstools, whereby bases with coverage depth <4 were reported as Ns[19]. Coverage statistics were then calculated usingseqtk comp, version 1.3[20].High-coverage samples with >65% genome coverage at depth >4 were retained[21].
Complex variant sites (MNPs) were decomposed into allelic primitives (SNPs and indels) and sites with zero non-reference alleles (allele counts, AC=0)within the high-coverage samples were removed using VCFlib commandsvcfallelicprimitivesandvcffilter -f"AC>0", respectively[18].
PHYLIP-format SNP representations were generated from the high-coverage VCF usingvcf2phylip, version 2.3[22]. A Washington sample was designated as the outgroup. DNA distance matrices were calculated using PHYLIP, version 3.697, byphylip dnadistwith default settings[23]. Unrooted trees were constructed by neighbor-joining usingphylip neighborwith random seed set to 133. Phenograms were generated withphylip drawgram.
This work was performed under an emergency order by the Chief Medical Officer of the Division of Public and Behavioral Health for the State of Nevada.The patient described herein provided written consent to publish this body of work.
Results
From March 1, 2020 through May 15, 2020, 19 431 specimens were tested by real-time PCR at the Nevada State Public Health Laboratory, of which 1264 specimens were deemed positive. By analysis of N1 gene detection by real-time PCR data, the average Ct value and standard deviation of such specimens was found to be 27.55±6.11. Of these positive cases, 23 were followed and were found to be associated with cases with no symptoms. The mean Ct value and standard deviation of these 23 cases was found to be 29.63±3.81. Of the 1264 positive specimens, 30 were from cases that involved COVID-19 related fatality of the tested patient. The average Ct value and standard deviation of these 30 fatal cases was 23.36±5.73. The difference between mean Ct values of fatal cases and all cases (4.19) was considered statistically significant(P=0.0004, two-tailed test) as well as the differences between the means of fatal cases and asymptomatic cases (6.27) (P=0.0004). However, the difference in mean Ct values between asymptomatic cases and all cases (2.08) may not be (P=0.103) (summary of mean values inTable 1).
For each collected specimen, the real-time PCR test to assess for the presence/absence of analyte (SARSCoV-2) RNA includes a co-analysis for detection of human RNase P RNA (RP). Mean RP Ct values and standard deviations of all cases, asymptomatic cases,and fatal cases respectively were 26.11±2.29,26.35±2.29 and 24.79±2.62. The differences of 1.32 between the mean Ct values of fatal cases and of all cases was deemed statistically significant according to two-tailed test (P=0.006). Observing this, we sought to determine whether decreased RP Ct values demonstrated a correlation to decreased analyte (N1)Ct values for specimens overall. We calculated the coefficient of determination (R2) for N1 Ct valuesvs.RP Ct values and found it to be 0.0187 (n=300 consecutively tested specimens), indicating a very weak relationship between the two values overall.
With regard to the differences in the amount of viral genomic material associated with swabs based on disease outcome, we sought to determine whether there are any genetic differences in the viruses associated with fatalitiesvs.those generally detected.We performed sequencing on the virions associated with 16 of 33 cases which involved fatality (selected at random), and compared these sequences to sequences generated from 154 other cases selected randomly from positive cases from multiple locations throughout Nevada. The sequences of virus were assessed for the presence of polymorphisms that correlated with disease severity or Ct value.
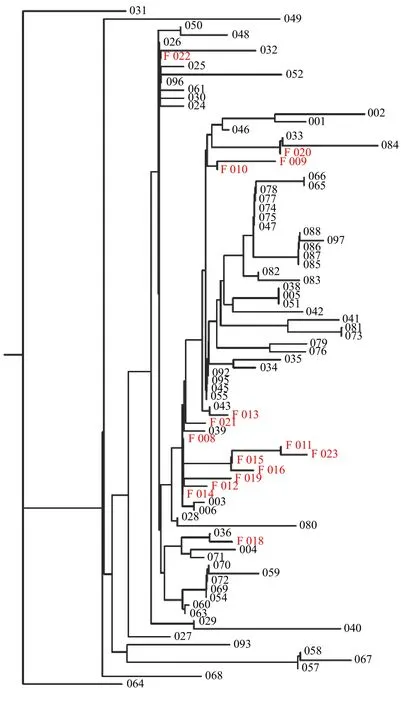
Fig. 1 Dendrogram of fatal cases, in relation to non-fatal.
Virus associated with fatality or low Ct showed a variety of genotype, with no singular strain / sequence associated with all such cases. Only two mutations were found that were exclusively associated with fatal cases. As shown inFig. 1, one branch of epidemiologically linked cases showed only fatal cases (011, 015, 016, and 023). The four cases were deaths that occurred within a 2-week period among residents of the same senior living community. These cases were associated with low Ct value in three of four instances (17.60, 18.35, 16.16, and 29.47). All four cases had two base changes relative to the reference sequence that were not seen in any of 156 other cases sequenced (97 shown). The first change was at base 18377 (C>T) which results in a change of an alanine to a threonine at amino acid position 6038 of theorf1abpolyprotein gene. This location denotes the reading frame of a 3′-to-5′ RNA exonuclease. The second change seen among these cases is at position 28187 (T>C) which results in a change of leucine to serine at amino acid position 95 in theorf8gene. The alteration at base 18377 was seen in four other sequences submitted to nextstrain.org out of 3104 total genomic sequences submitted as of June 20,2020. The T>C base change seen at 28187 was not previously observed according to nextstrain.org. Each of the four cases harbored virus that had the D614G alteration as well, which has been associated with higher infectivity and poorer outcomes[3–6]. All four cases involved people over the age of 77 at the same long-term living facility.
Discussion
An increased viral load is a logical correlate to poor disease outcome. It is measured quantitatively in the case of hepatitis B, C and HIV not only for the reason to monitor pharmacological efficacy but also to monitor potential disease progression. In the case of SARS-CoV-2, the observation herein matches this phenomenology with regard to COVID-19 based fatality. It is of note that in addition to viral analyte,(statistically) significantly more RNase P target was detectable in swabs from fatal cases than those found generally. There are many potential reasons hypothesized for this. Fatal cases may be associated with great inflammation, such that swabbed areas contain more cells of immune origin. Infection of cells in the swabbed areas may lead to greater cell death,and release of cellular RNA/DNA. Perhaps such patients are in a physical state that facilitates more significant physical swabbing (e.g., unconscious or deceased) than could take place on a healthier patient.Whatever the reason, might it be that the difference in the amount of viral genome seen in fatal and general swabs is a result of this difference in specimen quality/quantity? A difference of 4.19 Ct values (seen between fatal cases and general cases) on a theoretical standard curve with a PCR efficiency of 1.0 would correspond to an 18.2-fold difference in the amount of detected genome. Assuming a PCR efficiency for detection of RNase P of 1.0, and using a theoretical standard curve the difference of 1.32 Ct for RP would be expected to correspond to a 2.5-fold difference in the amount of collected human RNA on the swab.This would seem to imply that the difference in specimen collection likely does not account for the large difference seen for viral genome, unless the relationship between RNase P collection and viral genome collection is for some reason non-linear. The coefficient of determination (R2) between analyte (N1)and RP Ct values showed extremely weak correlation.This finding would seem to reject a hypothesis that the difference in the amount of viral RNA detected in specimens from fatal casesvs.non-fatal cases is caused by discordant sampling.
To date, no data has been generated to indicate that certain versions/strains of SARS-CoV-2 are more virulent than others. There is evidence that certain genotypes correlate with enhanced success in transmissibility among humans (e.g., D614G). The virulence of a genotype may be contextual to a host,complicating the ability to ascertain whether certain strains are potentially more dangerous. Herein, two base-pair changes were identified that were found in virions associated with four fatalities exclusively. The polymorphisms are extremely rare in the global database (nextstrain.org), thusly and any association with fatality outside of the cases herein is non-existent at present. It is noteworthy that one change, A6038T,causes a change in the 3′-to-5′ exonuclease protein.Mutagenesis of the gene for this protein (nsp14) has been shown in coronavirus to modulate the virus'pathogenicity and its ability to evade host immunity[24–25]. This is of interest in consideration of recent findings associated withorf8, the other gene found modified in these four fatal cases. Theorf8gene has been shown also to modulate immune evasion through MHC class I downregulation[26]. Whether either of these mutations played a biological role in the fatalities with which they were associated is unclear. All four fatality-associated viral genomes also harbored D614G, which has been associated with poor outcome[6]. These cases are epidemiologically related and included people of advanced age. More sequencebased surveillance data will be needed before any strict associations could be made.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- An unusual COVID-19 case with over four months of viral shedding in the presence of low neutralizing antibodies: a case report
- Identification of therapeutic drugs against COVID-19 through computational investigation on drug repurposing and structural modification
- Subgroup comparison of COVID-19 case and mortality with associated factors in Mississippi: findings from analysis of the first four months of public data
- Identification of county-level health factors associated with COVID-19 mortality in the United States
- Modeling the transmission dynamics of COVID-19 epidemic: a systematic review
- Challenges confronting rural hospitals accentuated during COVID-19