CRISPR/Cas9技术在作物中的研究及应用进展
2021-01-04罗银,刘峰
罗 银,刘 峰
(湖南大学研究生院隆平分院/湖南省蔬菜研究所,长沙 410125)
随着时代的发展,DNA测序技术(DNA sequencing)正不断进步与完善,水稻、玉米、番茄等多种作物已完成基因组测序[1~3]。如今科研工作者们将目光更多地集中在该怎么去解读大量的基因组信息、功能以及该如何对基因组进行精准而又高效的修饰,因此当基因组编辑技术问世时便举世瞩目。基因组编辑技术(Genome editing)是一种借助序列特异核酸酶(Sequence-specific nucleases,SSNs)对目标基因进行精确定点修饰的技术,可以使靶位点的碱基发生替换、缺失、插入等的改变。目前SSNs主要有4种类型,即归巢核酸酶(Mega nucleases,MNs)、锌指核酸酶(Zinc finger nucleases,ZFNs)、类转录激活因子效应物核酸酶(Transcription activator-like effect or nucleases,TALENs)和成簇的规律间隔的短回文重复序列及其相关系统(Clustered regularly interspaced short palindromic repeats/CRISPR-associated 9,CRISPR/Cas9 system)[4~12]。这些SSNs可以在靶位点切割序列,产生DNA双链断裂(Double strand breaks,DSBs),激活细胞内的两种DNA损伤修复途径:非同源末端连接(Non-homologous ending-joining,NHEJ)及同源重组(Homologous recombination,HR)[10],从而实现精确的基因组编辑。本文对CRISPR/Cas9的基本概况、优化及其应用等方面进行阐述。
1 CRISPR/Cas9的基本概况
1.1 基本结构与作用原理
CRISPR/Cas系统的组成部分是CRISPR序列与Cas基因家族。CRISPR序列由高度保守的顺向重复序列和长度相近的间隔序列组成,在第1个重复序列的上游存在一个先导序列(leader sequence),它是CRISPR序列转录的启动子,转录产生的非编码RNA(CRISPR RNAs,crRNAs)与Cas蛋白一起参与细胞内CRISPR/Cas系统的免疫作用。科学家们在CRISPR序列位点附近发现了CRISPR相关基因(CRISPR-associated gene,Cas gene),并发现其核心元件序列不一样,因此,学者们将CRISPR/Cas系统分为Ⅰ、Ⅱ、Ⅲ型(表1)[13]。Cas蛋白具有核酸酶功能,可以对DNA序列进行特异性切割,在Ⅰ型、Ⅲ型系统中,负责切割的是由多个Cas蛋白形成的复合物,在Ⅱ型系统中只用1个Cas9蛋白可切割DNA双链。Cas9蛋白有两个结构域:具有氨基末端的RuvC-like及HNH核酸酶结构域,每个结构域都与DNA的一条链结合,在单向导RNA(Single guide RNA,sgRNA)的引导下,通过对前间隔序列临近基序(protospacer adjacent motif,PAM)的识别来靶定目标DNA序列并对其进行切割,形成DSBs。

表1 CRISPR/Cas系统的类型Table 1 The types of CRISPR/Cas system
CRISPR/Cas系统源于细菌和古生菌的适应性防御,其免疫应答反应分为3个阶段。第一是获得阶段,噬菌体与外源DNA进入细菌后被识别结合到CRISPR阵列中产生免疫记忆后进入第二阶段——表达阶段,Cas蛋白被表达,CRISPR阵列被转录产生crRNA前体(Pre CRISPR RNA,Pre-crRNA),PrecrRNA经过反式激活CRISPR RNA(Trans-activating crRNA,tracrRNA)和核糖核酸酶Ⅲ(RibonucleaseⅢ,RNaseⅢ)加工成为成熟的crRNA,最后是干扰阶段,当噬菌体再次入侵时,CRISPR/Cas系统在crRNA的引导下Cas蛋白对噬菌体的基因组和质粒基因组切割形成DSBs。
最初,CRISPR/Cas系统是由Cas蛋白、crRNA和tracrRNA三部分构成,主要由crRNA对靶位点进行特异性识别。2012年,Jinek等[14]报道了Ⅱ型系统切割DNA双链的机制,对CRISPR/Cas9系统进行了优化,使crRNA与tracrRNA整合成一条sgRNA,由此CRISPR/Cas9系统只有sgRNA、Cas9蛋白两部分。在基因组编辑中,sgRNA与Cas9蛋白结合后与PAM序列进行匹配,从而识别目标基因序列,随后Cas9蛋白对靶序列进行切割,形成DSBs,诱发细胞内的NHEJ或HR途径对DSBs进行修复,高效地对基因组进行定点编辑与修饰(图1)。
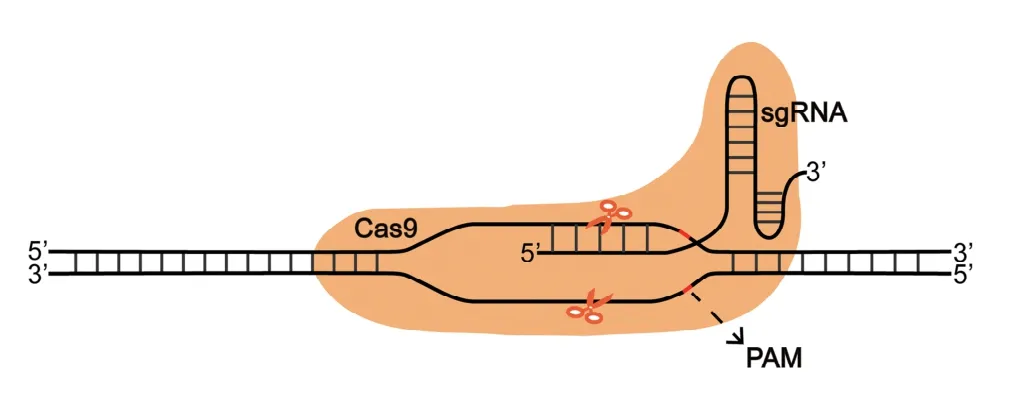
图1 CRISPR/Cas9系统模式图Fig.1 CRISPR/Cas9 system pattern diagram
1.2 发展历史
CRISPR/Cas系统始于1987年CRISPR序列的发现。1987年,Ishino等[15]科学家在E.coli中发现串联间隔重复序列,Horvath等[16]在2002年对嗜热链球菌全基因序列测序后提出了CRISPR的概念,然而那时他们并不了解它的功能,猜测它可能与噬菌体感染的免疫反应有关。到2007年,Barrangou等人[17]最先证明CRISPR序列确实能够抵御外来噬菌体的入侵。随着研究的深入,CRISPR系统的神秘面纱慢慢被揭开。Jinek等[14]在2012年取得重大进展,他们将crRNA与tracrRNA整合后形成一条sgRNA以切割DNA,使CRISPR/Cas9系统的构建更加简单,到此CRISPR/Cas9系统基本成型[18]。2013年Cong等[19]利用特异性RNA将Cas9带到靶位点进行切割,再加上Mali等[20]发现CRISPR/Cas9经过改造后可对人类细胞进行切割,能使目标基因发生定点突变、定点插入外源基因,基因组的定向编辑技术进一步发展,同年11月张锋与珍妮佛·杜德娜联合创建了Editas Medicine(EDIT)公司,CRISPR/Cas技术开始走向市场。2014年,比尔·盖茨基金会投资Editas Medicine公司,从此开始了CRISPR研究高潮。
1.3 修复DSBs的途径
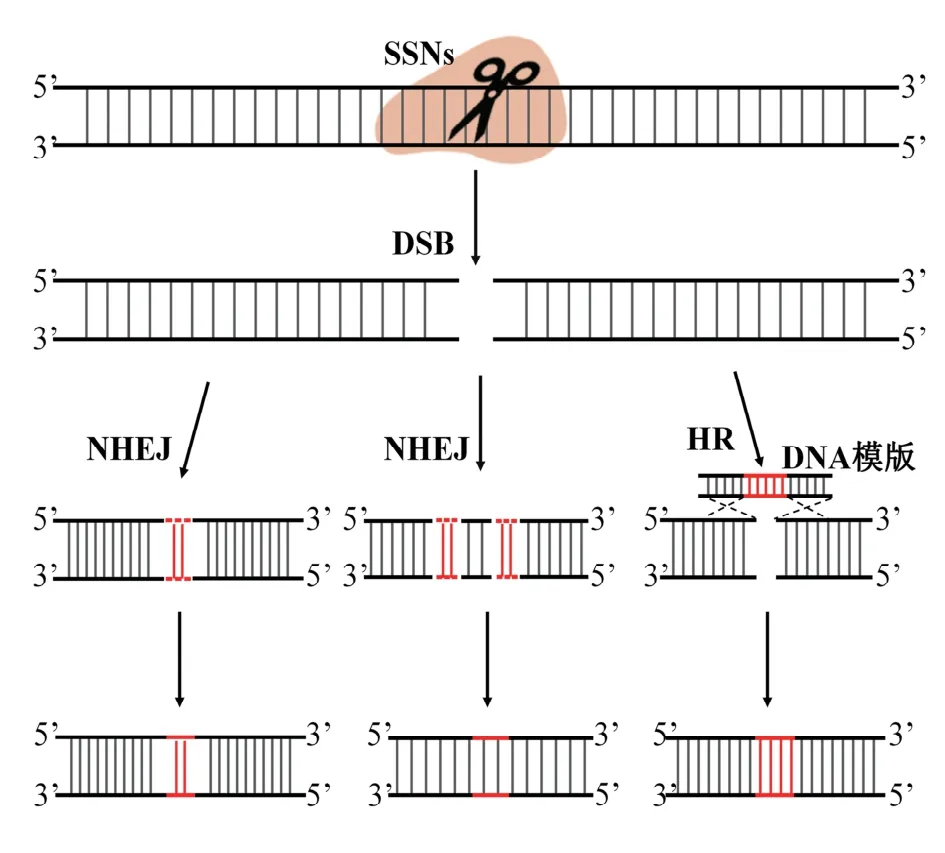
图2 DNA修复途径Fig.2 DNA repair pathway
修复DSBs途径主要有两种:NHEJ和HR(图2)。在发生频率上,发生频率高的是NHEJ途径,基本上所有的细胞及G1、S、G2期都能发生,而HR途径的发生频率极低,主要在S、G2期[21];但在编辑精确度上,HR途径比NHEJ途径高。通过NHEJ途径,DNA双链重新黏合,在DSB处会有少量的核苷酸(Nucleotide)插入或删除(Insertions/Deletions,Indels)。在人工提供同源序列时,修复DSBs便是HR途径,它以同源序列为模板,会产生精确的定点替换或插入突变体[22]。
1.4 CRISPR/Cas9技术的优势
目前通过传统杂交育种进行遗传改良周期长,需大量的材料;物理或化学诱变虽能产生大量的突变位点,但很难掌握突变的方向,导致鉴定不易;RNAi能够使基因沉默,但是不能稳定遗传。与这些方法相比,基因组编辑技术的优势在于:(1)操作简单,成本较低,适用于任何物种,目前已在酵母、果蝇、人类细胞、拟南芥、烟草、玉米、小麦、水稻等多种模式动、植物中成功应用;(2)效率高,不需要像杂交育种那么长的周期;(3)精确度高,通过设计sgRNA便能特异性识别目标基因的序列,对任意靶位点进行编辑,而T-DNA插入突变等技术则很难从具有特殊结构的基因中获得突变体;(4)可通过聚合不同基因至同一区段,使基因能够稳定表达;(5)可同时进行多基因编辑,突变不同的靶位点时只用重新构建与靶序列互补的sgRNA,将其与CAS9亚克隆于同一转化载体中即可,大大提高了基因编辑的效率。
2 CRISPR/Cas9技术的优化
2.1 核心元件的优化
CRISPR/Cas9与ZFN、TALEN技术都存在着脱靶效应,目前对已经完成全基因组测序的作物可以通过在线工具(CasOFFinder、E-CRISPR、CRISPR-P等)预测发生脱靶的位点来选择合适的技术(或sgRNA)对目标基因进行编辑(表2),但还有很多作物的基因组并未完全测序,而且对于某些全基因组中不预测的脱靶效应还不能通过目前的在线工具进行预测[23]。
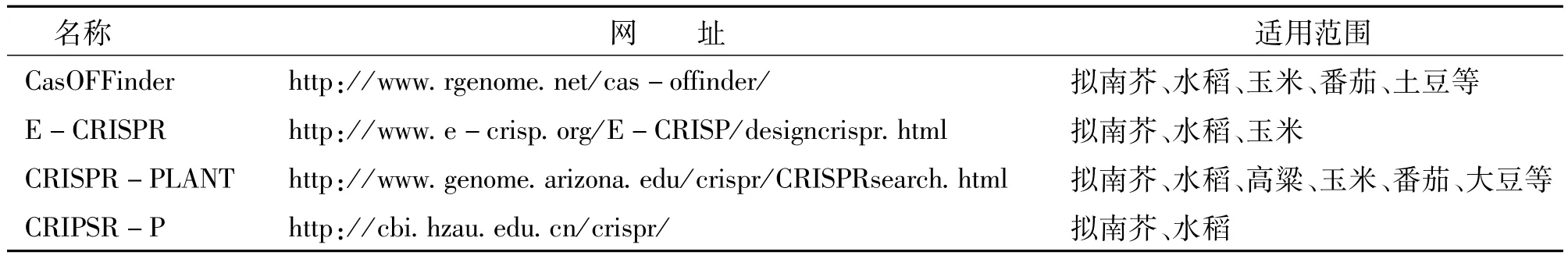
表2 适用于CRISPR/Cas9技术的在线工具Table 2 The online tools for CRISPR/Cas9 Technology
2.1.1 Cas蛋白的优化
对于Cas蛋白而言,Cas9蛋白识别PAM序列为NGG,限制了编辑范围。针对这点,科学家们在其他生物中进行了Cas蛋白的改造以扩大识别范围。当RuvC结构域上的氨基酸发生突变时,如第10位天冬氨酸(Aspartic acid,Asp)突变为丙氨酸(Alanine,Ala),会导致结构域失活,不具有切割功能,于是Cas9蛋白只有切口酶的功能,只能切割一条与HNH结构域结合的链,当HNH结构域上的氨基酸发生突变时,如第840位的组氨酸(Histidine,His)突变成丙氨酸(Alanine,Ala),结构域同样会失活,这时能切割的就是与RuvC结构域结合的一条链,可高效介导外源基因的定点插入,大大降低了NHEJ途径带来的风险;当两个结构域都发生氨基酸突变时,Cas9蛋白就会变成失活的Cas9蛋白(dead Cas9,dCas9),能通过连接激活子或抑制子对基因进行表达调控。经过多年的研究,发现很多SpCas9同源蛋白,如识别“NNAGAA”PAM序列的嗜热乳链球菌Cas9核酸酶(St1Cas9)[23~25]、识别“NNGRRT”PAM序列的金黄色葡萄球菌Cas9核酸酶(SaCas9)[26],扩大基因组编辑的范围。Cas12a蛋白,即Cpf1,它与Cas9蛋白相比区别在于Cas9蛋白切割形成平末端,Cas12a蛋白切割形成粘性末端,有4~5 nt的粘性凸起,使植物更易以HR方式对DSBs进行精确的替换,Cas12a蛋白的sgRNA更短,提高了Cas12a的打靶效率,且它的脱靶率更低,识别序列时TTTN,有利于对AT含量较高的DNA区域进行打靶。Cas13a蛋白可以在sgRNA引导下识别RNA并进行切割。
2.1.2 sgRNA的优化
对于sgRNA而言,理论上较长的靶DNA序列会降低脱靶的概率,然而从Fu等[27]的研究来看,17~19 nt长的sgRNA比20 nt的sgRNA活性更强、脱靶率更低,当sgRNA缩短为15 nt时则会失活。2017年Kweon等[28]通过开发融合向导RNA(fgRNA)与Cas9和Cas12a蛋白共同作用以减轻脱靶效应,而fgRNA并不会降低编辑效率。
2.1.3 Cas9蛋白与sgRNA同时优化
对Cas9蛋白与sgRNA同时进行优化,即基因组编辑技术的新成员——先导编辑(Prime Editing),其同时具有搜索和替换的功能,能够介导靶向的插入、缺失和替换。该技术通过将Cas9切口酶(SpCas9 H840A)与反转录酶(Reverse Transcriptase)融合表达形成Prime Editor(PE),通过使用经改造过的gRNA——导向编辑指导RNA(Prime editing guide RNA,pegRNA)实现对目标基因的编辑。
2.1.4 启动子的选择
对于启动子而言,在构建CRISPR载体时通常使用组成型启动子来启动Cas蛋白的表达,如35S,但此类启动子只会造成体细胞基因的编辑,使编辑效率低,因此科研人员研究不同的启动子,包括在生殖细胞、细胞分裂活跃的组织中特异性表达的启动子(表3)。

表3 启动Cas蛋白表达的启动子Table 3 Promoters that activate the expression of Cas protein
2.2 单碱基编辑系统
NHEJ修复途径发生频率高,但其修复不精确,通常会产生少量的Indels产物,而HR修复途径虽然比NHEJ途径精确度高,但是细胞类型、细胞周期对HR途径的限制很大,而且这两条途径相互竞争,导致HR途径很难高效地产生稳定的单碱基突变。在2016年由David Liu课题组开发了一种新的基因组编辑技术——碱基编辑技术(Base editing),实现了对基因组的精准编辑,使CRISPR/Cas能够对特定碱基进行修改[29,30]。该技术以CRISPR/Cas系统为基础,通过dCas蛋白或nCas蛋白与作用于单链DNA的脱氨酶结合来实现对靶位点的碱基替换,根据碱基修饰酶的类型分为胞嘧啶碱基编辑器(cytosine base editor,CBE)与腺嘌呤碱基编辑器(adenine base editor,ABE)两种,CBE系统与ABE系统分别利用胞嘧啶脱氨酶(Cytosine deaminase,CD)、腺嘌呤脱氨酶(Adenine deaminase,AD)能够实现C-T或A-G的碱基替换(图3)。单碱基编辑系统的优势主要表现在三个方面:(1)不需要引入DSBs。BE技术只需dCas蛋白或nCas蛋白即可实现对碱基定点替换;(2)与HR途径相比,BE系统不需提供模板DNA。HR途径必须在存在同源序列的情况下才能进行基因的定点编辑,最重要的是目前很难统一供体DNA的形式以及同源序列的长度,对于该怎么将足够的供体DNA有效地运送到细胞中这一难题也尚未完全解决[31],因此HR途径有很多不确定性。但BE系统可直接利用CD或AD通过sgRNA的引导高效实现C-T或A-G的替换;(3)BE系统的编辑效率高,适用于多种物种上。目前BE系统已经在动物细胞、人类细胞、植物、细菌等中被广泛使用[32~35]。
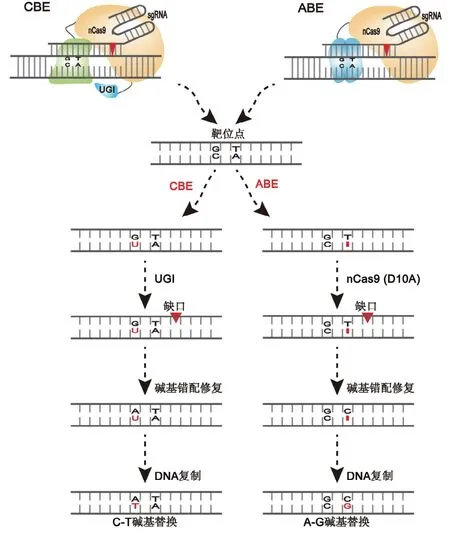
图3 ABE与CBE系统的工作原理Fig.3 Mechanismsof ABE and CBE system
3 CRISPR/Cas技术在作物中的应用
3.1 水稻
针对水稻基因的功能及品种改良科学家们进行了很多研究,已有很大的进展。2013年,Shan等[36]通过利用经过优化后的Cas9进行原生质体的转化,发现OsPDS基因的突变率为14.5%~20.0%,Os-BAHD2、Os02g23823与OsMPK2发生Indels的比例在26.5%~38.0%,接着对OsPDS-SP1基因进行打靶,用基因枪法对其胚性愈伤组织进行转化,共获得96株转基因植株,其中有9株突变植株,突变率为9.4%,在这9株植株中有3株植株是发生双等位基因突变的且具有矮化、白化的特征。Xu等[37]利用两个35S启动子与U6-26启动子分别驱动SpCAS9与sgRNA,针对水稻BEL利用根癌农杆菌侵染胚性愈伤组织,获得90株转基因植株,其中14例有插入和缺失突变,纯合突变体呈现对苯达松敏感的表型。Li等[38]利用CRISPR/Cas9技术特异诱导水稻中Gn1a、DEP1、GS3与IPA1四种基因发生突变,这些基因分别在籽粒数、穗结构、粒大小与株型等发挥调控作用。对T0代转基因植株的表型与效率分析表明,该系统在诱导目标基因编辑方面具有很高的效率,分别为27.5%(IPA1)、42.5%(Gn1a)、57.5%(GS3)、67.5%(DEP1),Gn1a、GS3与DEP1突变体的T2代分别具有粒数增加、粒径增大、直立穗密集的特点。
3.2 小麦
2014年,Wang等[39]利用TALENs和CRISPR/Cas9系统对小麦内源基因进行改造,以获得新的能稳定遗传的性状,他们选择了3个MLO位点(TaMLO-A1、TaMLO-b1、TaMLO-d1),为修改3个TaMLO使用了一对TALENs(T-MLO),从T0代450株转基因植株中发现27株突变植株,突变效率为6.0%。接着利用CRISPR/Cas9系统在单个TaMLO等位基因中诱导突变,使用T7内切酶Ⅰ(T7E1)鉴定sgMLO-A1在小麦原生质体、植株中诱导的突变,在T0代的72株转基因植株中筛选出4个独立的突变体,突变效率为5.6%,与TALENs获得的突变效率相似,而且他们还发现这3个基因都参与了面包小麦对Bgt感染的反应,这3个同源等位基因同时突变使小麦对白粉病具有广谱抗性。2017年Zhang等[40]利用CRISPR/Cas9系统敲除TaEDR1基因得到了Taedr1突变体,利用PCRRE鉴定了5个突变株系(T0-1、T0-2、T0-3、T0-4、T0-5),测序结果表明T0-1、T0-2、T0-3三个基因组都存在移码突变,对207株T1植株进行PCRRE与测序分析,仅鉴定出5个纯合T1突变体(3个来自T0-2,2个来自T0-3)具有移码突变,与野生型相比,其白粉病微菌落少和易感性降低,由此可见其对白粉病的抗性增加。
3.3 番茄
2015年,由于缺乏有效的方法将DNA修复模板传递给植物细胞,使用HR途径修饰植物基因组一直是个挑战。ˇCermák等[41]使用双生病毒复制子对番茄基因组进行遗传修饰,其频率比传统的DNA传递方法(农杆菌)高10倍。他们在番茄的基因组中,在ANT1的启动子区和基因编码区之间设计了TALEN或CRISPR/Cas9位点,提供模板DNA,包括两个同源臂,即ANT1的启动子区和编码区,还添加了Nos的启动子区、抗生素基因的编码区、35S终止子区和35S启动子,因此TALEN或CRISPR进入基因组对其位点进行切割后启动植物HR修复机制,使模版DNA插入到基因组上,即在表达抗生素基因后还插入了一个35S强启动子基因,使原本由内源启动子启动的ANT1变成由35S启动,增加了ANT1的表达,得到花青素含量高的果实。2017年,为了研究SlMAPK3基因对干旱胁迫的响应,Wang等[42]使用CRISPR/Cas9系统敲除该基因获得slmapk3突变体,在干旱胁迫下,与野生型相比其萎蔫更严重、更高的H2O2含量、更低抗氧化酶活性以及更多的膜损伤,此外,敲除基因导致干旱胁迫应答基因Sl-LOX、SlGST、SlDREB的表达上调或下调,证明Sl-MAPK3通过保护细胞膜免受氧化损伤和调节胁迫相关基因的转录参与番茄植株的干旱反应,具有抵抗干旱的能力。2018年,Li等[43]利用CRISPR/Cas9技术对5个基因,即在SP编码区、SP5G编码区、CLV3启动子区、WUS顺式调控元件及GGP1开放阅读框上游进行编辑,这5个基因分别调控植株的株型、开花时间、果实大小及维生素C含量,将理想性状导入4份耐逆野生番茄材料中,发现驯化后的番茄植株株型变得紧凑,开花时间提前,果实变大、维生素C含量上升,且保持了亲本的抗病性与耐盐性。
3.4 黄瓜
2016年,Chandrasekaran等[44]利用CRISPR/Cas9技术对黄瓜的病毒抗性进行研究,以elF4E基因的N0与C0末端为靶位点构建了Cas9/sgRNA载体对该基因进行敲除,结果表明纯合T3代植株对黄瓜叶脉黄化病毒感染具有免疫力,对西葫芦黄化花叶病毒、木瓜环斑镶嵌病毒具有抗性,而杂合突变体与非突变体对这些病毒高度敏感,说明elF4E基因在黄瓜中具有广谱的抗病毒能力。2017年,戚晶晶等[45]利用CRISPR/Cas9技术敲除了CsVFB1基因,经单克隆测序后在21个有效单克隆中检测到4个突变,成功获得转基因矮生黄瓜T0代植株,突变效率为19.05%。2018年,杨丽等[46]对CRISPR/Cas9载体进行优化,对CsVFB1、CsMLO8、CsGAD1基因进行编辑,发现T0植株编辑效率为100%,提高了黄瓜CRISPR/Cas9系统的编辑效率。利用CRISPR/Cas9系统,将CsWIP1基因敲除,从后代中筛选出纯合突变体Cswip1,使雌雄同株转化为全雌株,创制了黄瓜全雌系种质材料。
4 展望
自SSNs被发明,在这十多年的时间里基因组编辑技术发生了翻天覆地的变化,CRISPR/Cas9技术极大地加快了对作物基因功能的研究速度,同时也能作为一种新型育种技术(Newbreedingtechniques,NBTs)对作物品种进行改良,然而CRISPR/Cas9技术还面临着很多问题亟待解决。
首先是脱靶问题。CRISPR/Cas9技术在植物中发生的脱靶可以通过后代的分离而消除,若能消除脱靶,对其发展则有十分深远的影响。本文中提到了多种方法可以提高其特异性,比如对Cas蛋白进行优化等。
第二,提高编辑效率。首先,针对DNA修复途径来提高编辑效率,不同物种、细胞类型发生HR途径频率有很大的区别。(1)细胞周期的同步化。HR途径主要发生在S、G2期,NHEJ途径主要发生在G1、S、G2期,细胞周期不同所选择的修复方式也不同,可通过对细胞周期同步化后再进行基因组编辑。(2)供体DNA的优化。通过优化供体DNA的数量、长度、类型、导入方式,可提高HR途径效率。如Baltes等[47]利用烟草为研究材料,使用双生病毒载体表达SSNs与DNA模板,与T-DNA载体相比,HR途径效率有了很大提高。其次,针对CRISPR/Cas9系统进行优化,如核心元件的优化。
最后,各国政府对基因编辑作物的监管问题。随着CRISPR/Cas9技术的发展,越来越多的学者利用其进行品种改良,能够将需要几年甚至是几十年的时间缩短至几个月,更重要的是CRISPR/Cas9技术能够获得理想的特性并将其稳定遗传给后代,但是这样所获得的基因编辑作物会给世界带来什么样的结果还一无所知,人们对此十分关注。目前各国政府对转基因生物(Genetically modified organisms,GMOs)主要有两种监管态度[48]:(1)以欧盟为代表的基于过程的监管(Process-based),基因重组技术被认为具有潜在的危险性。无论通过这项技术获得什么基因或生物,只要有外源DNA转入,哪怕最终体内不存在外源DNA,都需要进行安全性评估与监测;(2)以美国为代表的基于产品的监管(Productbased),认为GMOs与非转基因生物(Non-genetically modified organisms,Non-GMOs)之间没有本质区别,需要监管的是生物技术的产物,而不是技术本身。相比较而言,过程监管要比产品监管更严格。美国农业监管机构认为,基因编辑作物(Genome edited crops,GECs)是通过激活细胞中的DNA修复机制而产生的突变,类似于自然突变、物理化学诱导突变,不属于GMOs。2016年,杨亦农教授通过CRISPR/Cas9技术培育的白蘑菇经过批准可不受监管,成为第一个脱离监管的GEC。到目前为止,美国农业监管机构已经确定了至少5种GECs不属于GMOs,包括ZFNs技术创造的低植酸玉米品种、TALEN技术创造的耐冷藏马铃薯品种及高油酸、低亚油酸大豆,以及CRISPR/Cas9技术创造的抗褐化双孢菇和抗除草剂玉米[49]。由于受多种因素的影响,欧盟对待GMOs的态度十分谨慎,对待转基因及基因编辑等NBTs的态度也十分保守,导致其产业格局以及产品的开发落后于美国等国家。中国国务院发布的《农业转基因生物安全管理条例》明确指出,农业转基因生物是指利用基因工程技术改变基因组构成,用于农业生产或农产品加工的动植物、微生物及其产品。然而目前对于GECs的监管标准没有明确的规定,对其的监管态度也没有相关的报道。尽管基因组编辑技术只出现十几年,但对农业、医学的影响十分重大,目前全球基因组编辑技术产业格局逐渐成形,我国基因组编辑技术位于世界前列,针对我国国情及基因组编辑技术研究进展制定合理的法规对正确引导我国基因组编辑技术的发展与应用至关重要。
综上所述,CRISPR/Cas9技术在基因功能分析、植物分子育种、人类疾病靶向治疗等方面具有无限潜力,虽然存在着一些问题,但是随着更多研究人员对该技术深入研究会使其逐渐完善,希望我国能在夯实现有基础上再向前一步,形成我国基因组编辑技术应用的优势领域,鼓励创新,发展具有自主知识产权的基因组编辑技术,争取在该领域中的主动权和话语权。
