“三台”核桃不同成熟期果实转录组分析
2020-12-28贺娜耿树香宁德鲁肖良俊
贺娜 耿树香 宁德鲁 肖良俊


摘要 以云南优良品种“三台”核桃4个不同成熟期种仁为材料,从分子水平上探索云南核桃(Juglans sigillata)种子不同成熟期种仁合成相关基因的表达模式。共有53 868条unigenes得到了注释,其中33 407条unigenes注释在Nr数据库中;31 725条unigenes注释在KEGG数据库中;Swissprot数据库中注释到19 842条unigenes;COG数据库中注释到16 655条unigenes。四大数据库共注释到33 753条unigenes,占总unigenes的62.66%。CK-vs-HT1、CK-vs-HT2、CK-vs-HT3、HT1-vs-HT2、HT1-vs-HT3、HT2-vs-HT3分别有 2 417、2 703、4 166、1 256、5 347、3 283条上调表达的差异基因和9 515、12 455、9 498、1 751、3 132、1 365条下调表达的差异基因。不同时期基因差异表达富集度最高的为脂肪酸生物合成代谢通路。核桃果实在成熟中后期,油脂及蛋白大量积累,相关基因表達量增加。
关键词 “三台”核桃 ;成熟度;转录组;基因
中图分类号 S 664.1 文献标识码 A 文章编号 0517-6611(2020)23-0165-05
doi:10.3969/j.issn.0517-6611.2020.23.041
Transcriptome Analysis of Santai Walnut Fruits at Different Ripening Stages
HE Na, GENG Shu-xiang, NING De-lu et al
(Yunnan Academy of Forestry and Grassland Sciences,Kunming, Yunnan 650201)
Abstract The expression patterns of genes related to seed synthesis at different mature stages of Juglans sigillata were explored at the molecular level by using 4 different mature kernels of Santai walnut as materials. A total of 53 868 unigenes were annotated, of which 33 407 were in the NR database, 31 725 in the KEGG database, 19 842 in the Swissprot database, and 16 655 in the COG database. A total of 33 753 unigenes were annotated in the four databases, accounting for 62.66% of the total. There were 2 417, 2 703, 4 166, 1 256, 5 347, 3 283 up regulated genes and 9 515, 1 2455, 9 498, 1 751, 3 132, 1 365 down regulated genes in CK-vs-HT1, CK-vs-HT2, CK-vs-HT3, HT1-vs-HT2, HT1-vs-HT3 and HT2-vs-HT3, respectively. Fatty acid biosynthesis pathway was the highest gene expression enrichment in different stages. In the middle and late stage of walnut fruit ripening, a large amount of oil and protein were accumulated, and the expression of related genes was increased.
Key words Santai walnut;Maturity;Transcriptome;Gene
云南“三台”核桃是云南众多深纹核桃(Juglans sigillata)中的一个主要栽培品种,产于云南宾川县、大姚县三台乡等地,无性系品种,脂肪含量44.42%~72.74%,蛋白质含量17.26%[1]。普通核桃(Juglans regia)为二倍体植物(2n=32),全基因组约606 Mb[2]。核桃是药食兼用植物,随高通量测序技术的飞速发展,基于高通量测序的转录组分析成为核桃代谢组研究的有力工具。转录组学是核桃功能基因组学一个重要分支,在发现核桃次生代谢产物生物合成关键基因、阐明次生代谢调控、筛选分子标记等方面具有重要的应用价值[3]。核桃代谢产物的研究主要集中于普通核桃脂肪代谢机制的分子生物学研究上,利用转录组测序技术构建普通核桃花后60、90和120 d脂肪积累期胚组织的转录组、表达谱,同时分析核桃胚脂肪积累期的基因表达模式及其代谢通路及核桃胚脂肪积累期脂肪代谢相关基因的表达模式,并利用转录组数据,克隆核桃异质型乙酰辅酶A羧化酶People acetyl coenzyme A carboxylase(ACCase)4个亚基的基因ORF[4-5]。
项目在前期的研究中发现云南“三台”核桃4个不同成熟期中的代谢产物含量差异较大,特别是次生代谢产物角鲨烯及磷脂类。选择4个不同成熟期“三台”核桃种仁构建云南核桃不同成熟期转录组文库,并利用生物信息学方法对转录的基因结构进行分析,对不同发育阶段差异表达基因进行GO功能富集分析,初步阐明不同成熟期云南核桃不同发育阶段差异表达基因功能及转录水平。
1 材料与方法
1.1 植物材料
以云南主栽核桃 “三台”为对象,依据方文亮等[1]对云南核桃果实成熟动态的研究结果和Li等[6]对果实发育动态的研究结果,分别在所选3株“三台”核桃样树树冠中部东南西北4个方位随机采摘30个果实,立取种仁置于液氮中,于-80 ℃冰箱保存,备用。4个成熟期分别为硬核期(6月CK样)、油脂转化期(7月HT1样)、油脂积累期(8月HT2样)、果实成熟期(9月HT3样)。
1.2 主要设备
冷冻高速离心机、核酸定量仪、-80 ℃超低温冰箱(德国Thermo公司)、全自动凝胶图像分析系统、水平电泳仪(美国 Bio-rad公司)、PCR扩增仪(美国ABI公司)、NanoPhotometer spectrophotometer、QuantStudio 12K Flex Real-Time PCR System(美国Thermo Fisher公司)、全套Eppendorf微量移液器(德国Eppendorf公司)、Agilent 2100 Bioanalyzer(Agilent RNA 6000 Nano Kit)。
1.3 主要试剂
Trizol reagent(Promega,Madison,USA)、NanoPhotometer spectrophotometer(IMPLEN,CA,USA)、NEBNext Ultra RNA Library Prep Kit for Illumina(Illumina Inc,San Diego,USA)、TruSeq PE Cluster Kit v3-cBot-HS(Illumia,San Diego,USA)、 SYBR Premix Ex Taq(TaKaRa,RR420)、DNA Marker-DL2000(TaKaRa 公司)、PCR 2×mix(上海碧云天生物技术有限公司)、SYBR Green Realtime PCR Master Mix(日本 Toyobo)。
1.4 试验及数据处理方法
1.4.1 “三台”核桃果实RNA的提取。
使用Trizol试剂提取“三台”核桃果实总RNA20 μg,再用1%琼脂糖凝胶电泳检测RNA是否污染及降解,用Agilent2100 Bioanalyzer检测总 RNA的浓度、RIN值等。使用Nano Drop超微量紫外分光光度计进行总RNA纯度检测。
1.4.2 cDNA文库构建和测序。
“三台”核桃的转录组测序由广州基迪奥生物科技有限公司完成。构建流程如下:通过带有Oligo(dT)的磁珠富集具有polyA尾巴的真核mRNA后,用超声波把mRNA打断。以片段化的mRNA为模板,随机寡核苷酸为引物,在M-MuLV逆转录酶体系中合成cDNA第一条链,随后用RNase H降解RNA链,并在DNA polymerase I 体系下,以dNTPs为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复、加A尾并连接测序接头,用AMPure XP beads筛选200 bp左右的cDNA,进行PCR扩增并再次使用AMPure XP beads纯化PCR产物,最终获得文库[7-8]。
1.4.3 测序数据的组装。
经测序仪产生的原始数据经base caling转化为序列数据,称为raw reads,结果以fastq文件存储,某些raw reads 带有adaptor序列,含有少量低质量序列包括N比例大于10%的reads、质量值Q≤5的碱基数占整个read的50%以上,经数据处理,得clean reads。使用短reads组装软件Trinity重头组装,先将具有一定长度overlap的reads连成更长片断,通过reads overlap 关系得到的组装片断为Contig,再将reads比对回Contig,通过paired-end reads 确定来自同一转录本不同Contig及Contig之间的距离,Trinity将这些Contig 连一起,得到两端不能再延长的序列称为Unigene,使用Tgicl去冗余和拼接,再对这些序列进行同源转录本聚类,得最终Unigene[9],该Unigene分为两部分,一是clusters(CL开头),另一是singletons(以Unigene开头),再将Unigene序列与Blastx比对,取最好比对结果蛋白确定Unigene序列方向。
1.4.4 转录组基因功能注释和表达分析。
为了获得基因功能注释,将所得的unigenes与NCBI的Nt数据库(NCBI non-redundant protein sequences database)、Nr数据库(NCBI nucleotide sequences database)、KOG 数据库(euKaryotic Ortholog Groups)以及Swiss-Prot数据库等进行比对,过滤 E-value 的阈值设置为较低 1×10-5。在获得所有 unigenes 的GO注释信息后,使用WEGO软件进行GO功能分类。此外,使用Blastx,过滤 E-value 的阈值设置为1×10-10,将所有unigenes與KEGG数据(the Kyoto Encyclopedia of Genes and Genomes Pathway Database)进行比对,分析核桃相关的代谢途径,挖掘与核桃次生代谢产物合成相关的基因。
1.4.5 基因差异表达分析。
使用 Bowtie 2[10]将 clean reads 比对到参考序列以统计基因比对率,之后再使用 RSEM[11]计算基因和转录本的表达水平。RSEM是用于转录组reads计算基因以及转录本表达量的软件包。RSEM 用 Paired-end的关系、reads的长度、fragment的长度分布、质量值等,基于最大期望的算法建立最大似然的丰度估计模型,用以区分哪些转录本是同一个基因的不同亚型。表达定量的结果以FPKM 为单位。差异表达基因(differentially expressed gene,DEG)检测基于泊松分布。为了提高 DEG的准确性,将差异倍数为2倍以上并且 Q-value≤0.001 的基因定义为显著差异表达基因。
2 结果与分析
2.1 转录组质量评估
使用 BGISEQ-500平台进行测序,测序结果如表1所示,共得到 7 306 Mb 的原始数据,48 707 124条raw reads。通过去除低质量 reads,获得48 647 921条clean reads,且clean reads的Q20比例为97.71%,GC含量为42.91%。以上数据证明本次测序质量较好,可用于后续拼接与分析。利用转录组拼接软件 Trinity对clean reads进行从头拼接,拼接获得的 unigene 数目为53 868个,总长度为 52 736 772 bp,平均长度为979 bp,N50长度为1 710 bp。组装结果质量评估可从N50来评估。将所有unigene从长到短排序,并依次累加长度,当累加片段长度达到总片段长度(所有unigene的长度)的50%时,对应那个片段的长度和数量,即为unigene N50长度和数量。unigene N50越长,数量越少,说明组装质量越好[12]。此项分析统计结果中包含了N50、GC含量等详细统计结果(表1)。
2.2 基因注释与分类
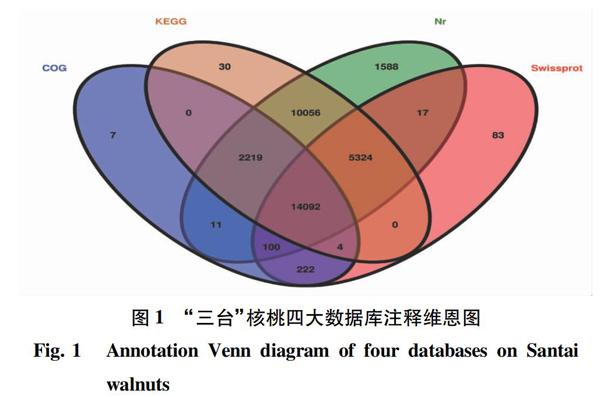
利用本地版Blast将获得的unigenes与四大功能数据库 Nr、KEGG、Swissprot、GO/COG进行比对,注释情况见图1 。共有53 868条unigenes得到了注释,其中33 407条unigenes注释在Nr数据库中;31 725条unigenes注释在KEGG数据库中;Swissprot数据库中注释到19 842条unigenes;COG数据库中注释到16 655 unigenes。四大数据库共注释到33 753条unigenes,占总unigenes的62.66%。此外,仍有20 115条unigenes未获得注释信息,可能是由于数据库中缺乏相关注释信息或者为新的转录本。
2.3 Nr注释分析
Nr数据库属于非冗余蛋白序列数据库NR(non-redundant protein sequence database)非冗余蛋白库,NCBI收录,所有GenBank+EMBL+DDBJ+PDB中的非冗余蛋白序列,对于所有已知的或可能的编码序列,NR记录中都给出了相应的氨基酸序列(通过已知或可能的读码框推断而来)以及专门蛋白数据库中的序列号[13]。“三台”核桃序列与Nr数据库比对的结果如图2。其中有25 505条unigenes比对到云南核桃即铁核桃上,基因配比率达76.34%,其余依次为土瓶草(2.46%)、可可(2.12%)、葡萄(1.14%)、木豆(1.01%)、黄麻(0.85%)、粉掌(0.76%)、枣(0.74%)、乌梅(0.61%)等。而其余13.37%的序列没有明确匹配到相应物种,可能是因为分散到了多种其他物种,或由于产生了一些数据库里没有的新基因。
2.4 KEGG pathway分析
基于 KEGG pathway的分析有助于进一步了解核桃基因的生物学功能。“三台”核桃的53 868条unigenes中有31 725条注释到了KEGG数据库中(表2)。KEGG(kyoto encyclopedia of genes and genomes)的代谢途径主要分为新陈代谢(metabolism)、遗传信息过程(geneticInformation processing)、环境信息过程(environmental information processing)、细胞过程(cellular processes)和生物系统(organismal systems)等。其中,新陈代谢共注释到 6 662条unigenes(20.99%),遗传信息过程注释到 2 298条 unigenes(7.87%),环境信息过程注释到546条unigenes,细胞过程注释到567条 unigenes,生物系统注释到422条unigenes(图3)。
核桃的主要生物活性物質为脂类和蛋白,因此着重对新陈代谢(metabolism)分类下的二级分类进行分析。在KEGG注释中与新陈代谢相关的是global and overview maps(2 633条unigenes,39.52%),碳水化合代谢(936条unigenes,14.05%),能量代谢(589条unigenes,8.84%),氨基酸代谢(586条unigenes,8.79%),脂类代谢(510条unigenes,7.65%),
辅因子和维生素代谢(310条unigenes,4.65%),其他次级代谢生物合成(256条unigenes,3.84%),核苷酸代谢(254条unigenes,3.81%),其他氨基酸代谢(253条unigenes,3.79%),萜类及聚酮化合物代谢(189条unigenes,2.83%),多糖的生物合成和代谢(146条unigenes,2.19%)。这些注释将为进一步研究与核桃油脂及蛋白合成有关的代谢途径、基因的结构和功能提供参考信息。
2.5 COG功能分类
COG 是直系同源家族蛋白数据库,按照COG的分类信息将“三台”核桃的16 655条unigenes分配到 25个类别里,结果见图4。其中,最多的是一般功能预测,包含4 870条unigenes,占总unigenes的29.24%;其余有代表性的包括信号转导机制(2 260条 unigenes,13.56%),转录后修饰、蛋白转换、伴侣蛋白(1 889条 unigenes,11.34%),翻译(1 121条 unigenes,6.73%),翻译、核糖体构成和起源(960条 unigenes,5.76%),胞内运输、分泌及囊泡转运(917条 unigenes,5.50%),未知功能(903条 unigenes,5.42%),碳水化合物的运输和代谢(892条 unigenes,5.35%),RNA加工及修饰(784条 unigenes,4.70%),能量产生及转化(704条 unigenes,4.22%),次生代谢产物合成、运输及分解(685条 unigenes,4.11%)等,其中最少是细胞迁移(13条 unigenes,0.08%),其中未知功能(903条 unigenes,5.42%)表明“三台”核桃中存在许多新基因,其生物学功能有待挖掘。
2.6 不同成熟期核桃差异基因分析
将差异倍数为2倍以上并且 Q-value≤0.001 的基因确定为显著差异表达基因。对云南“三台”核桃4个不同成熟期样进行两两比较,进行差异基因分析(图5)。其中7月与6月相比有2 417个基因上调表达,9 515个基因下调表达;8月与6月相比有2 703个基因上调表达,12 455个基因下调表达;9月与6月相比有4 166个基因上调表达,9 498个基因下调表达;8月与7月相比有1 256个基因上调表达,1 751个基因下调表达;9月与7月相比有5 347个基因上调表达,3 132个基因下调表达;9月与8月相比有3 283个基因上调表达,1 365个基因下调表达。核桃果实在成熟中后期,油脂及蛋白大量积累,相关基因表达量增加。在核桃发育期,果实油脂积累期与硬核期相比,下调表达基因最多,也是差异基因较多的一组,这与CK样品有关,CK样品为果实整个子房,其他3个样品(HT1、HT2、HT3)均为果肉,另外,果实形成初期也是果实整个发育过程中代谢最活跃的时期,尤其是激素在子房中的含量,子房中几乎包括果实发育期间所有必需的生物代谢途径[14]。从HT1到HT3,后一个阶段都比前一阶段上调表达的基因多,这表明果实处于不断发育变化中。HT1和HT3上调基因比例明显增加,核桃发育最快,HT2和HT3相比差异基因减少,可能与后期果实成熟代谢减慢有关。
2.7 GO富集分析
Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表(controlled vocabulary)来全面描述生物体中基因和基因产物的属性。GO总共有3个ontology(本体),分别描述基因的分子功能(molecular function)、细胞组分(cellular component)、参与的生物过程(biological process)[15]。将不同成熟期“三台”核桃的差异基因按照其功能进行GO分类,将每个分类数量最多的2种term和数量列入图6中。在生物学过程(biological process)中,metabolic process(代谢过程)、cellular process(細胞过程)和single-organism process(单组织过程)所占的比例最多;细胞成分(cellular component)中,cell(细胞)、cell part(细胞部分)、organelle(细胞类脂质)所占的比例最多;分子功能(molecular function)中,数量最多的为catalytic activity(催化活性)和binding(结合)。
3 结论
(1)以4个不同成熟期(硬核期、油脂转化期、油脂积累期、果实成熟期)云南主栽的“三台”核桃为材料,转录组分析不同成熟期基因表达关系。共有53 868条unigenes得到了注释,其中33 407条unigenes注释在Nr数据库中;31 725条unigenes注释在KEGG数据库中;Swissprot数据库中注释到19 842条unigenes;COG数据库中注释到16 655条unigenes。四大数据库共注释到33 753条unigenes,占总unigenes的62.66%。
(2)不同成熟期“三台”核桃差异基因两两比较中,CK-vs-HT1、CK-vs-HT2、CK-vs-HT3、HT1-vs-HT2、HT1-vs-HT3、HT2-vs-HT3分别有 2 417、2 703、4 166、1 256、5 347、3 283条上调表达的差异基因和9 515、12 455、9 498、1 751、3 132、1 365条下调表达的差异基因。
(3)在“三台”核桃生物合成途径中,以次生代谢产物生物合成途径注释到的两端不能再延长的序列最多,在脂肪酸代谢途径中富集到的差异表达基因最多,其中acc系列酶及FAB系列基因可能是“三台”核桃油脂和脂肪酸生物合成关键基因,其后续通过表达载体构建和蛋白异源表达等对基因功能进行验证。
参考文献
[1] 方文亮,宁德鲁.云南核桃[M].北京:科学出版社,2019.
[2] YOU F M,DEAL K R,WANG J R,et al.Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms [J].BMC Genomics,2012,13:1-16.
[3] RAI A,KAMOCHI H,SUZUKI H,et al.De novo transcriptome assembly and characterization of nine tissues of Lonicera japonica to identify potential candidate genes involved in chlorogenic acid,luteolosides,and secoiridoid biosynthesis pathways[J].Journal of natural medicines,2017,71(1):1-15.
[4] 张楠.核桃胚脂肪积累期转录组分析[D].泰安:山东农业大学,2014.
[5] 杨丽,陈虹,潘存德,等.核桃种子油脂转化期转录组分析[J].果树学报,2017,34(9):1084-1094.
[6] LI Y T,MA S M,WANG Y F,et al.The dynamics of fat,protein and sugar metabolism during walnut(Juglans regia L.)fruit development [J].African journal of biotechnology,2012,11(5):1267-1276.
[7] 潘教文,李臻,王庆国,等.NaCl处理谷子萌发期种子的转录组学分析[J].中国农业科学,2019,52(22):3964-3976.
[8] 张坤.红地球葡萄延后栽培生育后期树体与果实的水分关系研究[D].兰州:甘肃农业大学,2018.
[9] 王燕.萝卜铅(Pb)胁迫响应的分子机制研究[D].南京:南京农业大学,2014.
[10] LANGMEAD B,SALZBERG S L.Fast gapped-read alignment with Bowtie 2[J].Nature methods,2012,9(4):357-359.
[11] LI B,DEWEY C N.RSEM:Accurate transcript quantification from RNA-Seq data with or without a reference genome[J].BMC Bioinformatics,2011,12:1-16.
[12] 鲁强.三桠苦种子休眠的解除方法和初步机理研究[D].广州: 广州中医药大学,2019.
[13] 刘文浩.熏硫及脱硫处理对龙眼贮藏生理的影响及转录组研究[D].广州:华南农业大学,2016.
[14] 李文彬.‘红阳猕猴桃果实发育转录组及花青素累积机理研究[D].武汉:中国科学院研究生院(武汉植物园),2015.
[15] 白琳云.灵武长枣嫁接与根蘖繁殖植株生长、果实特性的比较[D].银川:宁夏大学,2019.
基金项目 云南省重大专项科技计划项目(2018ZG003);云南省科技创新人才培养项目(2016HB004)。
作者简介 贺娜(1982—),女,湖北京山人,副研究员,硕士,从事经济林栽培及精深加工研究。
*通信作者,研究员,博士,从事植物资源评价及木本油料精深加工研究。
收稿日期 2020-05-07
