3,4-取代[1,2,4]三唑衍生物的合成及其表征
2020-12-24易绣光严云逸李雅静赖飞平
*易绣光,张 敏,严云逸,徐 瑶,李雅静,赖飞平
(1.井冈山大学化学化工学院,江西,吉安 343009; 2.国家电投集团新能源科技有限公司,江西,南昌,330096)
0 引言
三氮唑作为一类富含π 电子的芳香性氮杂环化合物,在抗炎[1]、抗菌[2]、抗病毒[3]、抗癌[4]、消炎[5]、抗结核[6]、除草[7]、杀虫[8]以及调节植物生长[9]等领域具有广泛的生物活性,因而一直是化学家及药物学家们关注的重点。
三氮唑是一个药物设计领域重要的活性片段,它的芳香性和富电子性质使其易于与各种不同的酶和受体结合,从而赋予其广泛的生物活性[10]。自20世纪60年代,由荷兰Philiph-Dupher 公司首次报道1,2,4-三氮唑类威菌灵杀菌剂及德国Bayer 公司和比利时Janssen 公司报道1-取代唑类衍生物杀菌活性以来[11-13],得到了迅猛发展。三氮唑类化合物的合成会受到广大科研工作者的重点关注,其主要原因在于:含有三个N 杂原子的三唑类化合物因其与生物体内的氢键形成息息相关,并且与生物环境具有良好的亲和性,在生物体内与靶蛋白发生相互作用从而导致活性因子的叠加,增强化合物的生物活性。
然而对近几年上市的三唑类化合物进行分析,发现虽然三唑类药物结构多样化,但基本上都是通过对其侧链部分的结构修饰而达到产生更强的生物活性。如,Shrestha 等人通过烷烃类侧链修饰得到的三氮唑醇类化合物[14]。Chandrika 等人通过烷氨类侧链修饰得到的具有烷基和氨基取代的三氮唑醇衍生物[15]。Chandrika 等人通过环胺类侧链修饰得到具有哌嗪类侧链衍生物[16]。Sadeghpour 等人通过杂环类侧链合成了含有3-硝基-氟康唑类似物[17]。Zou 等人通过硫类侧链合成了含二硫代氨基甲酸酯侧链化合物[18]。
通过上述分析表明,三唑类化合物通常是通过侧链结构的修饰,使其结构多样性、新颖性。目前为止上述烷(氨)基化等法主要是用烷(氨)基化试剂和三氮唑或其衍生物直接反应而得到理想结构分子。然而,其高温、高压,耗时长,后处理操作萃取、柱层析等苛刻的反应条件大大限制了其工业化生产,尤其是遇到一些结构复杂的化合物,空间位阻严重影响其产率等。正基于此,本研究通过利用3-溴基苯甲酸甲酯和3-氨基苯甲酸甲酯为原料,经过肼解、缩合、环合等反应,寻求一条反应条件温和、处理分离容易、更适合工业化生产的合成路线,从而得到四个新的1,2,4-三氮唑化合物,通过核磁共振谱测定其结构并通过高效液相色谱分析其纯度。
1 实验部分
1.1 主要仪器和试剂
1.1.1 主要试剂
3-溴苯甲酸甲酯(AR,国药试剂);3-氨基苯甲酸甲酯(AR,国药试剂);水合肼(AR,国药试剂);DMF-DMA(AR,国药试剂);异丙胺(AR,国药试剂);(R)-2-氨基-1-丙醇(AR,国药试剂)及一些常规溶剂。
1.1.2 主要仪器
Avance III 400MHz 核磁共振仪(德国Bruker公司);SUMMIT P680型高效液相色谱仪(美国DIONET 公司);DF-101S集热式恒温加热磁力搅拌器(巩义市予华仪器有限公司)等。
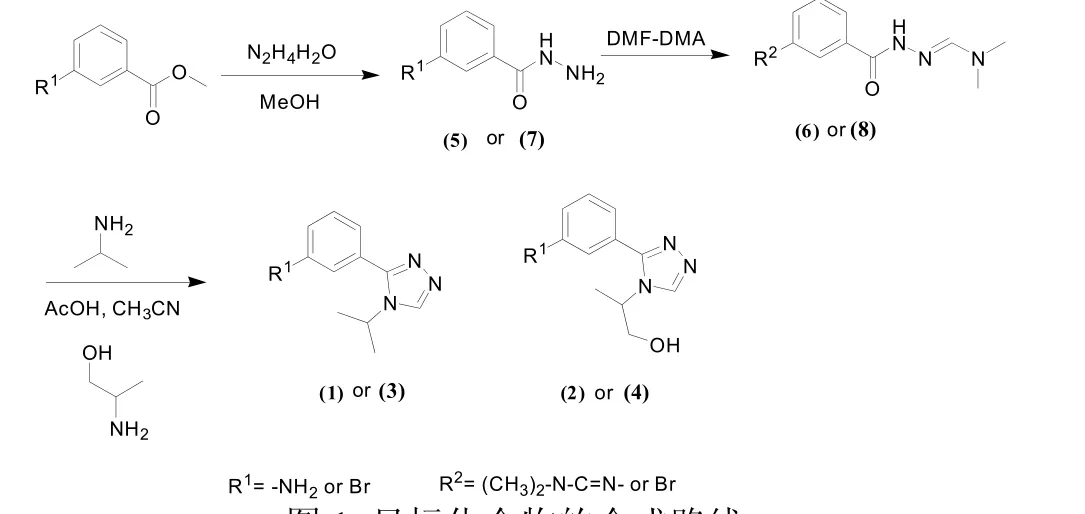
图1 目标化合物的合成路线Fig.1 The synthesis route of target compounds
1.2 实验过程
将3-氨基苯甲酸甲酯(100 g,0.66 mol)甲醇800 mL,90%的水合肼89.46 g,室温下搅拌2 h,有大量白色固体析出,过滤,洗涤,干燥得化合物(5):3-氨基苯酰肼80 g,收率80%。
将化合物(5)3-氨基苯酰肼(100 g,0.66 mol),N,N-二甲基甲酰胺二甲基缩醛(DMF-DMA 600 mL),加热回流3 h,薄层色谱板跟踪反应完成。减压除去DMF-DMA,得黄色固体,用乙酸乙酯洗涤,干燥得化合物(6):N’-[3-(二甲氨基亚甲肼羰基)-苯基]-N,N’-二甲基甲脒120.6 g,收率70%。
将化合物(6)(10 g,0.04 mol),乙腈20 mL,冰酸酸5 mL 及异丙胺11.30 g,充入氮气保护下加热回流12 h,薄层色谱板跟踪反应完成。减压浓缩除去溶剂,倒入冰水中,有大量白色固体析出,过滤,用乙酸乙酯洗涤,干燥得化合物(1):3-(3-氨基苯基)-4-异丙基-4H[1,2,4]-三唑5.2 g,收率:65%。
将化合物(6)(10 g,0.04 mol),乙腈20 mL,冰酸酸5 mL 及(R)-2-氨基-1-丙醇14.37 g,充入氮气保护下加热回流12 h,薄层色谱板跟踪反应完成。减压浓缩除去溶剂,倒入冰水中,有大量白色固体析出,过滤,用乙酸乙酯洗涤,干燥得化合物(2):3-(3-氨基苯基)-4-异丙醇基-4H- [1,2,4]-三唑6.3 g,收率:72%。
同理用3-溴苯甲酸酯取代3-氨基苯甲酸甲酯按上述相同的方法:将3-溴苯甲酸甲酯(141 g,0.66 mol),甲醇800 mL,90%的水合肼89.46 g,室温下搅拌2 h,有大量白色固体析出,过滤,洗涤,干燥得化合物(7):3-溴苯酰肼113 g,收率80%。
将化合物(7)3-溴苯酰肼(141 g,0.66 mol),DMF-DMA(N,N-二甲基甲酰胺二甲基缩醛)600 mL,加热回流3 h,薄层色谱板跟踪反应完成。减压除去DMF-DMA,得黄色固体,用乙酸乙酯洗涤,干燥得化合物(8):N’-[3-(二甲氨基亚甲肼羰基)-苯基]-N,N’-二甲基甲脒124.3 g,收率70%。
将化合物(8)(10.8 g,0.04 mol),乙腈20 mL,冰酸酸5 mL 及异丙胺11.30 g,充入氮气保护下加热回流12 h,薄层色谱板跟踪反应完成。减压浓缩除去溶剂,倒入冰水中,有大量白色固体析出,过滤,用乙酸乙酯洗涤,干燥得化合物(3):3-(3-溴苯基)-4-异丙基-4H-[1,2,4]-三唑6.9 g,收率:65%。
将化合物(8)(10.8 g,0.04 mol),乙腈20 mL,冰酸酸5 mL 及2-氨基-1-丙醇14.37 g,充入氮气保护下加热回流12 h,薄层色谱板跟踪反应完成。减压浓缩除去溶剂,倒入冰水中,有大量白色固体析出,过滤,用乙酸乙酯洗涤,干燥得化合物(4):3-(3-溴苯基)-4-异丙醇基-4H-[1,2,4]-三唑8.1 g,收率:72%。
2 结果与讨论
目标化合物的制备如下:首先,分别以3-氨基苯甲酸甲酯和3-溴基苯甲酸甲酯为原料,在甲醇为溶剂中加入水合肼进行先亲核加成再消除的肼解反应。其次,在上述产物中加入DMF-DMA,DMF-DMA 受到酰胺上的氨基亲核进攻,很快就脱去二甲胺,产生一个活泼性更强的试剂,最后与氨基发生缩合反应得到亚胺,从而对氨基进行保护。目前相关文献报道的保护试剂主要有磷酰基[19]、对甲苯磺酰[20]基等,在这些保护试剂中还存在着诸多的缺点,比如:使用磷酰基作为保护剂实验过程要求无水无氧等苛刻的条件且脱保护不完全,收率较低;使用对甲苯磺酰基作为保护剂实验过程副反应较多,后续处理复杂,需要过柱纯化。然而在本实验过程中采取使用DMF-DMA 作为保护剂,条件反应温和,无需过柱分离,有利于工业化生产。在亚胺中加入异丙胺或2-氨基-1-丙醇在乙腈用溶剂中回流,乙酸作酸化剂使得亚胺上的酰基质子化,进行关环得到四个目标化合物。
化合物(1)1H NMR (400 MHz, CDCl3):δ8.31(s,1H, ArH), 7.27 (dd,J= 9.0, 6.6 Hz,1H, ArH),6.95-6.71 (m, 3H, ArH), 4.53 (dt,J= 13.5, 6.7 Hz, 1H,CH), 3.87 (s, 2H, NH2), 1.47 (d,J= 6.8 Hz, 6H, CH3);
化合物(2)1H NMR (400 MHz, CDCl3):δ8.71(s, 1H, ArH), 7.15 (t,J= 7.8 Hz, 1H, ArH), 6.86-6.75(m,1H, ArH), 6.70 (dd,J= 7.8,1.8 Hz, 2H, ArH),5.34(s,3H,OH, NH2), 4.30(dd,J=12.1,6.8 Hz,1H, CH), 3.60 (qd,J= 11.3, 6.1Hz, 2H, CH2), 1.35 (d,J= 6.9 Hz, 3H, CH3);
化合物(3)1H NMR (400 MHz, CDCl3):δ8.33(s, 1H, ArH), 7.73 (t,J= 1.7 Hz, 1H, ArH), 7.64 (ddd,J= 8.0, 1.9, 1.1 Hz, 1H, ArH), 7.53-7.45 (m, 1H, ArH),7.38 (t,J= 7.9 Hz, 1H, ArH), 4.45 (dt,J= 13.5, 6.7 Hz, 1H, CH), 1.49 (d,J= 6.7 Hz, 6H, CH3);
化合物(4)1H NMR (400 MHz, CDCl3):δ8.23(s,1H, ArH), 7.65 (s,1H, ArH), 7.50 (dd,J= 35.8,7.9Hz, 2H, ArH), 7.24 (dd,J= 17.0, 9.1 Hz, 1H, ArH),5.93 (s, 1H, OH), 4.68-4.22 (m, 1H, CH), 3.95 (dd,J= 24.9, 8.1 Hz, 2H, CH2), 3.95 (d,J= 7.0 Hz, 3H,CH3)。
通过高效液相(HPLC)流动相:乙腈:水= 60:40;流速:1 μL/3 min;检测器: UV254 nm;规格:50 nm×2.1 nm。使用面积归一法分别检测目标化合物(1)~(4)的纯度为:99.9%,98.9%,99.37%,99.26%,如下图2所示。

图2 目标化合物的高效液相谱图Fig.2 The high performance liquid spectrum of target compounds
3 小结
为了避免无水无氧等苛刻的实验条件以及后续处理的复杂性,通过使用DMF-DMA 作为合成三氮唑的保护和脱反护试剂,规避副主物的色谱柱分离,寻求一条操作简便、反应温和、更适合工业化的生产路线,并成功制备了四种新的三氮唑化合物,其结构通过1H NMR 进行了表征。通过HPLC面积归一法分别测得目标化合物的纯度为:99.9%,98.9%,99.37%,99.26%。
