(S)-噻吗洛尔半水合物的合成及表征
2020-12-23周政虎马晓旭
周政虎,左 波,马晓旭,吴 莉*
1.武汉工程大学化学与环境工程学院,湖北 武汉430025;
2.武汉科福新药有限责任公司,湖北 武汉430000
噻吗洛尔是作用最强的β受体阻断药,其作用强度为普萘洛尔的8倍,其中S-噻吗洛尔其药物作用效率是R-噻吗洛尔的80~90倍。噻吗洛尔早期是美国默克研究实验室有限公司1982年开发(商品名BLOCADREN)主要用于原发性高血压病、心绞痛或心肌梗塞后的治疗,近年来发现本品尚有明显的降低眼内压的作用。对青光眼,特别是对于原发性开角型青光眼(滴眼,减少房水形成,不缩瞳,无调节痉挛)有良好的效果,优于传统的降眼压药,其特点为起效快,副作用小,耐受性好[1-3]。同时噻吗洛尔在治疗婴幼儿血管瘤和肉瘤有一定的治疗效果[4-6]。
Wasson等[7-8]最早报导了相关的合成方法,收率为27.67 %。该工艺会产生N-取代异构体,不易分离。Narina[9]以外消旋体环氧氯丙烷和3-氯-4-吗啉-1,2,5-噻二唑进行醚化缩合,中间体经活性钴手性拆分后,再与叔丁胺反应得到S-噻吗洛尔,总收率为28.84 %。该合成路线中间体的拆分分离较为繁琐,拆分试剂较为昂贵,不适用生产。王德才等[10]采用传统的酒石酸成盐拆分工艺(酒石酸成盐、重结晶、游离三步),拆分收率6.94%;Peralampi等[11]的发明专利采用甲苯、正己烷、水3种溶剂转晶得到S-噻吗洛尔半水合物,游离转晶收率为66.85 %。综合关于S-噻吗洛尔的合成的相关文献[12-14]报道,选取3-吗啉-4-氯-1,2,5-噻二唑为起始原料,经过水解反应、醚化取代反应、胺化成盐反应、游离转晶4个步骤合成S-噻吗洛尔半水合物。同时对每步反应工艺参数进行了优化;并对目标产物采用红外光谱法(infrared spectrosco⁃py,IS)、核磁共振氢谱(hydrogen nuclear magnetic resonance spectroscopy,1H-NMR)、核磁共振碳谱(carbon nuclear magnetic resonance spectroscopy,13C-NMR)、质谱(mass spectroscopy,MS)、热重分析仪法(thermo gravimetric analysis,TGA)、差示扫描热量法(differential scanning calorimetry,DSC)表征。
1 实验部分
1.1 化学试剂及仪器
3-吗啉-4-氯-1,2,5-噻二唑(太仓市茜泾化工有限公司);R-环氧氯丙烷(沈阳金久奇科技有限公司,化学纯度99.73 %,光学纯度99.80 %);叔丁胺(天津市大茂化学试剂厂);二氯甲烷、无水乙醇、二甲基亚砜、三乙胺、氢氧化钠、氯化钠、碳酸钠、无水硫酸钠、氯化钠、盐酸、马来酸(国药化学试剂有限公司)。
VERTEX 80傅立叶变换红外光谱仪、AscendTM核磁共振波谱仪(德国布鲁克);Pyrisl TGA、Diamond DSC(美国珀金埃尔默仪器有限公司);TRAP/XCT液质联用仪(安捷伦科技有限公司);EMPYREANX射线衍射仪(荷兰帕纳科公司)。
Agilent Technologies R60 infinity液相色谱仪;化学纯度检测柱子及流动相:十八烷基硅烷键合硅胶为填充剂(Agilent SB C18 4.6 mm×150 mm,5μm),流动相A为4.32 g/L辛烷磺酸钠溶液(调节醋酸pH至3.0),流动相B为甲醇,流速为1.5 mL/min,紫外检测波长为295 nm,柱温为35℃;光学纯度检测柱子及流动相:表面键合有喹啉衍生物的硅胶为填充剂的手性色谱柱(DAICEL CHIRALPAK 4 mm×150 mm,3μm),流动相A为含50 mmol/L的甲酸和25 mmol/L二乙胺的甲醇,流动相B为V乙腈∶V水=10∶90的溶液,流速为0.5 mL/min,紫外检测波长为298 nm,柱温为25℃。
1.2 S-噻吗洛尔半水合物的合成
S-噻吗洛尔半水合物是由起始原料3-吗啉-4-氯-1,2,5-噻二唑经氢氧化钠水解得到中间体1,再与R-环氧氯丙烷发生醚化反应得到中间体2,与叔丁胺进行胺化反应及马来酸成盐得到中间体3,最后经氢氧化钠游离及水转晶拆分制得,其详细的合成路线见图1所示。
1.2.1 中间体1(3-吗啉-4-羟基-1,2,5-噻二唑)的合成在10 L的玻璃反应瓶中加入4 L水,30℃下加入390 g(9.75 mol)氢氧化钠,加500 g(2.43 mol)3-吗啉-4-氯-1,2,5-噻二唑和500 mL二甲基亚砜,加料完毕,升温至100℃反应3~4 h,反应完毕,冷却至室温,用稀盐酸调节pH至1~2,抽滤,80℃干燥至恒重得到425 g(2.27 mol)白色固体中间体1,收率93.41%,高效液相色谱法(high performance liquid chromatography,HPLC)纯度99.65%。质谱(mass spectrum,MS)(正离子模式,m/z):188.67[M+H]+;核磁共振氢谱(1H-NMR)(400 MHz,CD⁃Cl3):δ3.43(s,4H),δ3.69(s,4H),δ12.85(s,1H)。
1.2.2 中间体2((R)-4-(4-(环氧乙烷-2-基甲氧基)-1,2,5-噻二唑-3-基)吗啉)的合成在5 L反应瓶中加入1.70 kg(18.37 mol)R-环氧氯丙烷、430 g(2.30 mol)中间体1和116 g(1.15 mol)三乙胺,升温至75℃反应6~7 h。反应完毕,减压蒸除溶剂得到棕红色油状物。在5 L反应瓶中加入2.7 L水、108 g(2.70 mol)氢氧化钠,搅拌溶解。30℃下将蒸馏油状物加入至碱液中并保温搅拌1 h,抽滤,滤饼再用2 L水打浆1 h,抽滤,60℃干燥至恒重得到332 g中间体2粗品。再经乙醇重结晶得到217 g(0.89 mol)中间体2。收率:38.84%,HPLC纯度:96.33 %。MS(正离子模式,m/z):244.74[M+H]+;H-NMR(400 MHz,DMSO-d6):δ2.73~2.93(d,J=80 Hz,2H),δ3.40~3.41(d,J=4 Hz,1H),δ3.53(s,4H),δ3.81~3.82(d,J=4 Hz,4H),δ4.23~4.27(m,J=16 Hz,1H),δ4.75~4.78(m,J=12 Hz,1H)。
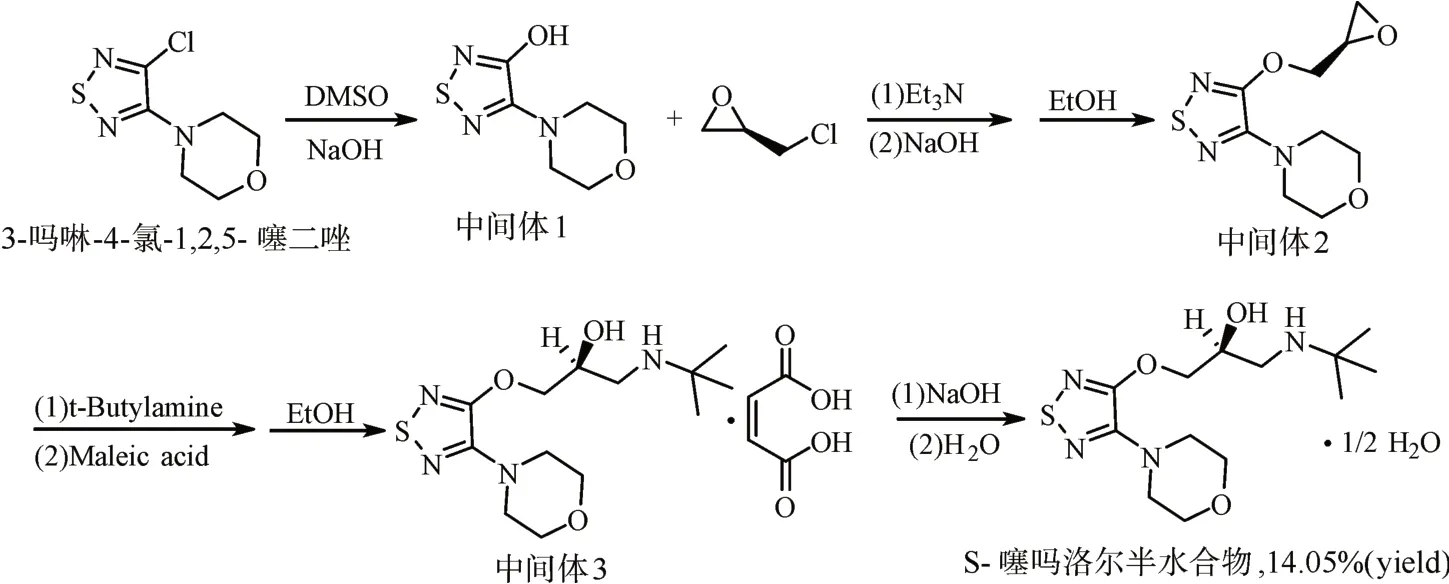
图1(S)-噻吗洛尔半水合物的合成Fig.1 Synthesis of(S)-timolol hemihydrate
1.2.3 中间体3(S-马来酸噻吗洛尔)的合成在5 L玻璃反应瓶中加入1.6 L无水乙醇、200 g(0.82 mol)中间体2和240 g(3.29 mol)叔丁胺,升温至45℃反应16 h,反应完毕,蒸除溶剂。将残留物中加入2 L水和400 mL二氯甲烷,调节浓盐酸pH至1~2,分层,水层再用400 mL二氯甲烷萃取1次。水相用氢氧化钠调节pH至13~14,用600 mL二氯甲烷萃取3次,合并有机相用600 mL水洗2次和600 mL质量分数10%氯化钠水溶液水洗1次,无水硫酸钠干燥,过滤,滤液减压浓缩蒸除溶剂,用1.6 L无水乙醇将浓缩后油状物溶解并转移至5 L反应瓶中升温至75℃,加入63 g(0.54 mol)马来酸,保温搅拌0.5 h。再降温至10℃抽滤,滤饼60℃干燥至恒重得到210 g(0.49 mol)中间体3粗品。进一步乙醇重结晶得到166 g(0.38 mol)白色固体中间体3。收率:46.73%。产物(HPLC)纯度为100 %,异构体(HPLC)为1.89%。MS(正离子模式,m/z):318.06[M+2H]+;1H-NMR(400 MHz,DMSO-d6):δ1.30(s,9H),δ2.51~2.52(d,J=4 Hz,2H),δ2.87~2.93(m,J=24 Hz,1H),δ3.13~3.16(d,J=12 Hz,1H),δ3.46(s,4H),δ3.71(s,4H),δ4.20(s,1H),δ4.34~4.40(m,J=24 Hz,2H),δ6.04(s,2H),δ8.40(s,2H)。
1.2.4 S-噻吗洛尔半水合物的合成在5 L反应瓶中加入1.6 L纯化水、160 g(0.37 mol)中间体3和500 mL二氯甲烷,用64 g(1.60 mol)氢氧化钠调节pH至13~14,分层,水层用500 mL二氯甲烷萃取2次,合并3次有机相,用500 mL的质量分数10%碳酸钠水溶液水洗1次,再用500 mL纯化水水洗3次,分层,无水硫酸钠干燥,过滤,滤液减压浓缩蒸除溶剂得到S-噻吗洛尔油状物。
在2 L反应瓶中,加入117 g(0.37 mol)S-噻吗洛尔油状物和600 mL纯化水25℃搅拌打浆14 h,抽滤,滤饼35℃鼓风干燥10 h,碾磨,将碾磨后固体用600 mL纯化水25℃下搅拌打浆2 h,抽滤,滤饼35℃鼓风干燥14 h至恒重得到100 g(0.31 mol)白色S-噻吗洛尔半水合物固体,收率82.87%,总收率为14.05%。产物纯度为99.97%(HPLC),异构体为0.17%(HPLC),e.e.值99.66%。熔点:49.6~50.5℃;MS(正离子模式,m/z):317.00[M+H]+;1H-NMR(400 MHz,CDCl3):δ4.48~4.37(m,J=44 Hz,2H),δ3.96~3.94(m,J=8 Hz,1H),δ3.81~3.79(m,J=12 Hz,4H),δ3.54~3.52(m,J=8 Hz,4H),δ2.81~2.78(m,J=12 Hz,1H),δ2.61~2.58(m,J=12 Hz,3H),δ1.11(s,9H);核 磁 共 振 碳 谱(13C-NMR)(100 MHz,CDCl3):δ153.80、149.93、72.86、68.16、6.48、47.91、44.36、29.05;红外 吸收 光 谱(IR):3 435.88、2 963.82、2 856.89、1 451.16、1 532.90、1 498.56、1 311.58、12 62.95、1 228.62、1 120.49 cm-1;DSC:尖锐吸热分解峰(42~52℃),最大吸收峰49.80℃;TGA:质量变化98.65%,热分解起始温度150℃,分解前失重2.64%。
2 结果与讨论
2.1 中间体2的工艺优化
2.1.1 缚酸剂类别对中间体2粗品的影响参考文献[15]用氢氧化钠、三乙胺合成普萘洛尔中间体。根据反应机理,设计用4种常用缚酸剂进行筛选。缚酸剂的优化结果见表1。以三乙胺或哌啶为缚酸剂的产物纯度和收率均比以氢氧化钠或碳酸氢钾条件高。三乙胺条件下纯度与哌啶条件下纯度无明显差别但收率明显高于后者,故选择三乙胺为第2步反应的缚酸剂。
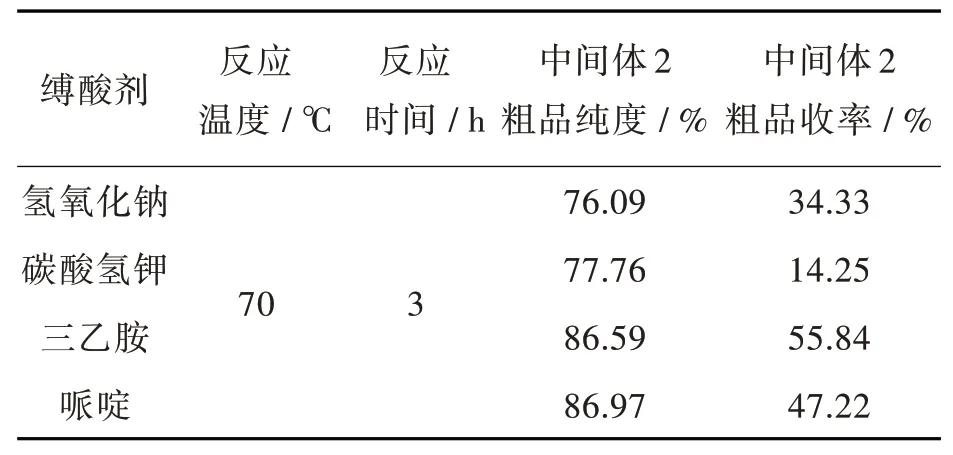
表1缚酸剂类别对醚化反应的影响Tab.1 Effects of acid binding agent on etherification reaction
2.1.2 反应温度对中间体2粗品的影响反应温度优化结果见表2。25、50、75℃下反应产物纯度相当且明显高于100℃;与25、50、75℃条件下三者对比,75℃的反应收率最高。综合纯度和收率2个因素,选择75℃为该步反应最佳温度。

表2反应温度对醚化反应影响Tab.2 Effects of reaction temperature on etherification reaction
2.2 中间体3的工艺优化
2.2.1 不同反应溶剂对胺化反应的影响反应溶剂优化实验结果见表3。由有关物质检测结果可知,二氯甲烷、四氢呋喃、叔丁胺3个条件下,反应液中还有大量原料未反应完;以乙醇为反应溶剂反应中原料已反应完全且反应液纯度最高(96.54 %)。综上所述,故选择乙醇为该步反应的溶剂。

表3反应溶剂类别对胺化反应影响Tab.3 Effects of reaction solvent on amination reaction
2.2.2 反应时间和反应温度对胺化反应的影响反应时间和反应温度优化结果见表4。同等反应时间下两温度对比:46℃下反应6 h剩余原料仅为5.04 %,而25℃下反应6 h仍有83.02 %原料未反应;故选择46℃为第3步反应温度。46℃条件下随着反应时间的增加,反应原料逐渐减少,反应液纯度逐渐增加,反应16 h反应原料已反应完全,且反应液纯度最高。故选择反应16 h为第3步反应时间。
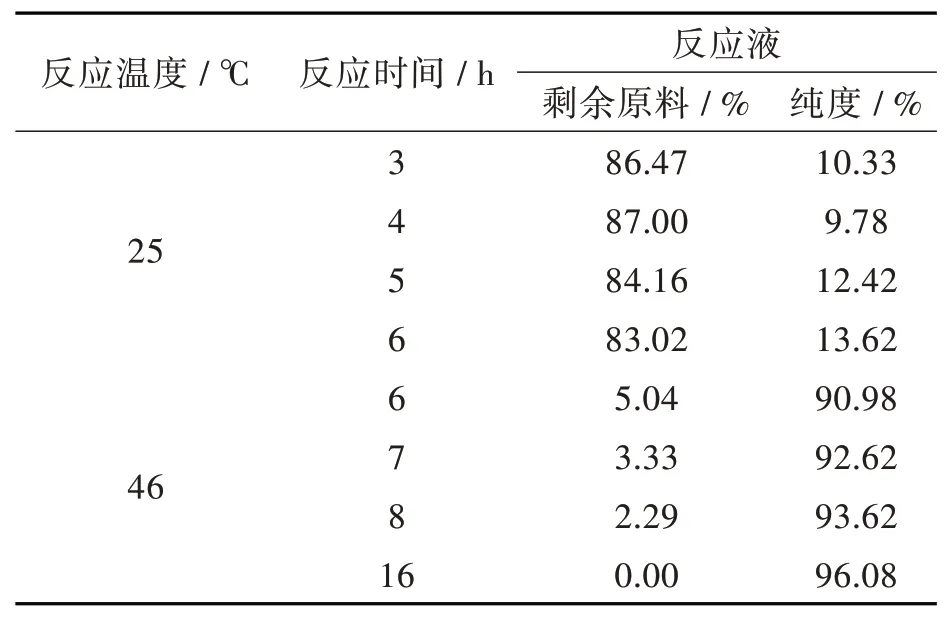
表4反应温度、时间对胺化反应影响Tab.4 Effects of reaction temperature and time on amination reaction
2.3 S-噻吗洛尔半水合物的工艺优化及表征
2.3.1 拆分工艺对成品的影响S-噻吗洛尔是由中间体3经过游离成S-噻吗洛尔油状物,而工艺采用R-环氧氯丙烷得到得中间体3异构体大于1.0%,故需要进行拆分研究。拆分工艺研究结果见表5。研究结果表明,采用酒石酸成盐和水打浆工艺,有关物质对比拆分前均变化不大,且两者相当。对比异构体,采用酒石酸成盐一次拆分异构体降低幅度不明显,从2.87%降低至2.42%;而采用水打浆成半水合物可以将异构体杂质从2.84%降低至0.54%,进而达到满足药典标准(≤1.0%)。故拆分工艺选择用水打浆制备成S-噻吗洛尔半水合物的工艺。
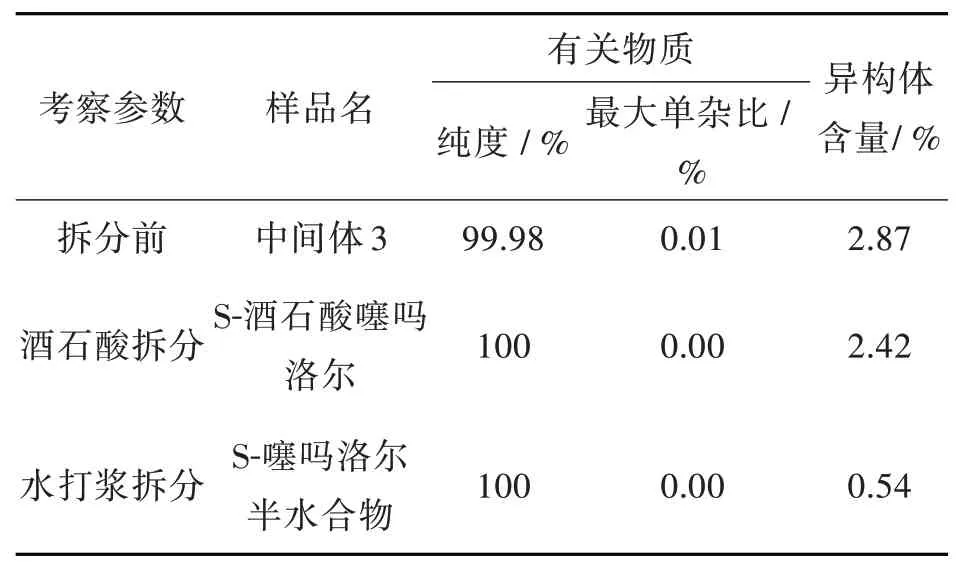
表5拆分工艺对成品的影响Tab.5 Impacts of separation process on final products
2.3.2 成品结晶半水的确证DSC[谱图见图2(a)]结果表明样品在42~52℃有一尖锐吸热分解峰,最大吸收峰49.80℃,表明该样品熔点在49.80℃,与实测值49.6~50.5℃一致;TGA[谱图见图2(b)]结果表明样品分解质量变化为98.65 %,热分解起始温度150℃,分解前失重2.64 %,该结果与S-噻吗洛尔半水合物中水的质量分数2.76%基本一致。
根据S-马来酸噻吗洛尔比旋度药典标准(CP2015)比旋度为-5.7°~-6.2°,由此推算S-噻吗洛尔半水合物的比旋度范围为(S-马来酸噻吗洛尔比旋度×噻吗洛尔半水合物分子量/马来酸噻吗洛尔分子量)-7.58°~-8.24°。样品比旋度为-8.02°,比旋度结果表明样品为S型,且结合DSC、TGA结果确定样品为半水合物。
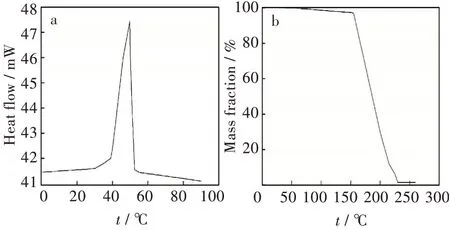
图2(S)-噻吗洛尔半水合物表征:(a)DSC曲线,(b)TGA曲线Fig.2 Characterization of(S)-timolol hemihydrate:(a)DSC curve,(b)TGA curve
3 结论
选择三乙胺为醚化反应缚酸剂时,最佳反应温度为75℃;选择乙醇为胺化反应的溶剂,46℃为该步反应温度,最佳反应时间16 h;S-噻吗洛尔的拆分工艺较传统的酒石酸成盐拆分工艺步骤缩短只需要水转晶拆分一步,且收率高;本文转晶拆分的溶剂单一(水作溶剂),且绿色、经济、环保。本文工艺合成的S-噻吗洛尔半水合物不同于S-噻吗洛尔成酸根,属于水合物,其物理属性及部分化学属性发生很大变化,属于新的化合物,在医药上有很大的应用前景,目前在国内还没有上市。
