基于机器学习方法预测环形RNA编码蛋白的潜能
2020-12-18刘晶晶宋晓峰
王 琮,赵 健,刘晶晶,宋晓峰
(南京航空航天大学 自动化学院,中国 南京 211106)
环形RNA的发现要追溯到20世纪70年代,1976年,人们首次在病毒中观察到环形RNA的存在[1].环形RNA起初被认为是转录过程中错误拼接而成的产物,是“转录垃圾”.随着二代测序技术的快速发展和生物信息学工具的开发,大量的环形RNA被发现,环形RNA成为转录组研究领域的新热点[2-3].2015年,Wang等[4]通过人工插入IRES序列构造环形RNA,并发现环形RNA能够翻译特定的绿色荧光蛋白,这引起了研究者们的重视.2017年,Ivano等[5]通过northern blot、质谱技术等方法验证了环形RNA(circ-ZNF609)能够编码蛋白质,从而调控肌细胞增殖.Yang等[6]通过抗体检测、质谱鉴定发现Circ-FBXW7能够翻译与恶性胶质瘤发病相关的蛋白FBXW7-185aa.Zhang等[7]验证了circ-SHPRH能够编码新型蛋白质SHPRH-146aa,该蛋白能够抑制神经胶质瘤的发生.2018年Zhang等[8]还验证了circ-PINT能够编码新型蛋白PINT87aa,抑制多种癌基因转录延伸.2019年Liang等[9]发现了circβ-catenin能够编码全新蛋白β-Catenin-370aa,调控Wnt/β连环蛋白信号通路.Heesch等人在心脏组织中发现40种环形RNA能够翻译.
越来越多的数据显示环形RNA能够编码蛋白质,在细胞生命活动中发挥至关重要的作用.本文基于环形RNA的序列与结构特征,计算环形RNA的开放阅读框(ORF),预测环形RNA的内部核糖体进入位点(IRES)和N6甲基化修饰位点(m6A),并构建机器学习模型预测环形RNA编码蛋白的潜能.
1 环形RNA序列与结构特征的计算
1.1 ORF的计算
开放阅读框(open reading frame,ORF)指的是从起始密码子(AUG)开始,结束于终止密码子(UAA, UAG, UGA)的一段连续的RNA片段,其中每3个碱基(或3联密码子)能够编码一种对应氨基酸.由于密码子起始位置的不同,RNA序列可能有3种开放阅读框(即Frame1, Frame2, Frame3).环形RNA编码蛋白的先决条件是具有一定长度的开放阅读框.然而现有计算开放阅读框的工具都是针对线性RNA开发的,环形RNA呈封闭环状结构,开放阅读框可能跨越反向剪接位点,出现长度超过环形RNA自身的情况.考虑到上述情况,我们自编程序计算环形RNA的开放阅读框,得出环形RNA中最大开放阅读框的位置及序列信息.
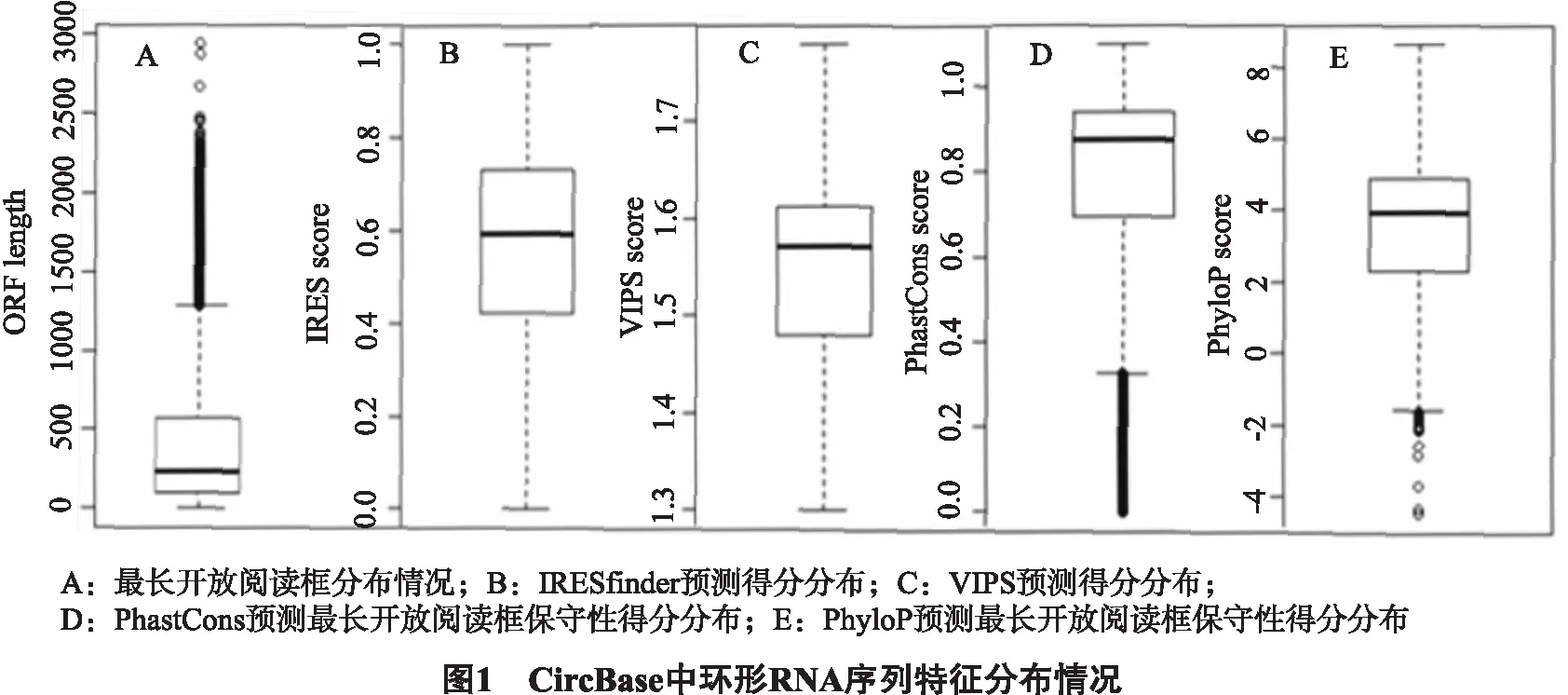
我们应用自编程序计算数据库CircBase中所有环形RNA的最大开放阅读框,其中剔除了长度大于 2 000 bp 的环形RNA.结果如图1A所示,经过统计得出,环形RNA开放阅读框中位数为 234 bp,大多分布在99~573 bp 之间,其中43%的环形RNA的开放阅读框大于 300 bp,具有编码超过 100 aa 长度的蛋白质的可能.
1.2 IRES的预测
RNA的翻译机制主要分为帽依赖起始机制和非帽依赖翻译起始机制2种.前者基于5’帽子结构,招募核糖体起始翻译,是大部分线性RNA翻译起始的方式.而后者从RNA内部起始翻译,需要一些特殊元件的介入.IRES(internal ribosomal entry Site)即内部核糖体进入位点,是RNA内部的一段序列.IRES能够直接招募核糖体亚基与之结合,从而启动翻译.由于环形RNA呈封闭环状结构,没有5’末端帽子,所以环形RNA的翻译只能通过非帽依赖的方式进行.IRES是非帽依赖翻译的必要元件,通过预测环形RNA中是否存在IRES,可判断环形RNA能否编码蛋白.
目前基于序列与结构特征预测IRES元件的生物信息学工具主要有IRESfinder[11]和VIPS[12].其中IRESfinder基于逻辑回归模型的方法,预测待测序列中是否存在IRES元件.而VIPS将待测序列与已知IRES进行局部结构比对,预测其是否具有IRES活性.IRESfinder与VIPS均被用于预测CircBase数据库中环形RNA是否存在IRES元件,结果如图1B、1C所示.在IRESfinder预测结果中,环形RNA具有IRES元件的得分中位数为0.592, 64.6%的环形RNA被预测存在IRES结构.VIPS预测得分中位数为1.57, 32.9%的环形RNA被预测存在IRES活性.对比2种IRES元件预测工具的结果,约20.3%的环形RNA被2种工具都预测为序列中存在IRES元件,如图2所示.

1.3 序列保守性的计算
RNA序列进化保守性指的是相似或相同的RNA序列出现在各物种间,该序列在物种进化过程中被保留下来.由于环形RNA是一类呈闭合环状结构的特殊RNA,特殊的结构决定了具有稳定性,它不被核糖核酸外切酶(RNase R)分解,相比线性RNA更为稳定.由环形RNA编码的蛋白也因此拥有更高的物种间的保守性.
PhastCons和Phylop被用于计算环形RNA的ORF区域、IRES区域、反向剪接位点附近区域)的进化保守性.首先,获取环形RNA指定区域在基因组上的坐标.接着,在UCSC下载人类基因组与其他物种多重序列比对结果文件(hg38.phastCons100way.bw和hg38.phyloP100way.bw).最后,使用BEDTools获取环形RNA指定区域的序列保守性情况.
CircBase数据库中所有环形RNA的最大开放阅读框区域的PhastCons和Phylop得分如图1D、1E所示.PhastCons预测得分的中位数为0.88,PhyloP预测得分的中位数为3.92,环形RNA的最大开放阅读框区域较为保守.
1.4 m6A修饰的预测
m6A,即N6甲基化修饰,腺苷酸第6号N在相关酶的催化下发生甲基化修饰事件.m6A是真核生物中最普遍的一种转录后修饰.有研究报道,环形RNA中富含m6A基序,而单个m6A位点足以驱动环形RNA起始翻译[13].
本文应用基于序列特征预测m6A修饰位点的生物信息学工具SRAMP[14],预测环形RNA中m6A修饰位点存在的可能性.CircBase数据库中环形RNA的m6A修饰得分情况和高置信度m6A修饰位点的个数如图3所示.m6A修饰预测得分的中位数为9.16,环形RNA中高置信度m6A修饰位点个数的平均值为1.65个,60.8%的环形RNA中具有至少一个高置信度m6A修饰位点.

1.5 其他特征的计算
目前转录本编码蛋白潜能预测工具主要有CPC[15]、CPAT[16]和CNCI[17],我们经过一定的预处理,将其应用于预测环形RNA编码蛋白潜能.利用上述3种生物信息学工具,计算环形RNA中的编码蛋白相关特征Fickett score、Hexamer score、PI等电点得分等.此外,还改进了k-mer的计算方法,以此提取环形RNA内部的序列特征.在改进的k-mer计算方法中,非末端的碱基可用X替代,X表示任意1种碱基.例如,在k=3时,序列AGTTCG可匹配的k-mer有AGT、AXT、GTT、GXT、TTC、TXC、TCG和TXG.考虑到计算量,只取k=1~5,共提取 2 500 种改进的k-mer特征.
2 预测模型的构建
利用机器学习方法,分别构建逻辑回归、支持向量机、随机森林和XGBoost 4种分类模型,以此预测环形RNA编码蛋白的潜能.
2.1 数据来源
正样本数据来源于4个部分,包括文献中实验验证的可编码蛋白的环形RNA、蛋白质质谱(MS)数据支撑的可编码蛋白的环形RNA、翻译组测序数据(Ribo-seq)验证的可编码蛋白的环形RNA和数据库circRNAdb[18]中注释为可编码蛋白环形RNA.以上4类数据经过去重复,共同组成正样本数据集,包含5 747个正样本数据.
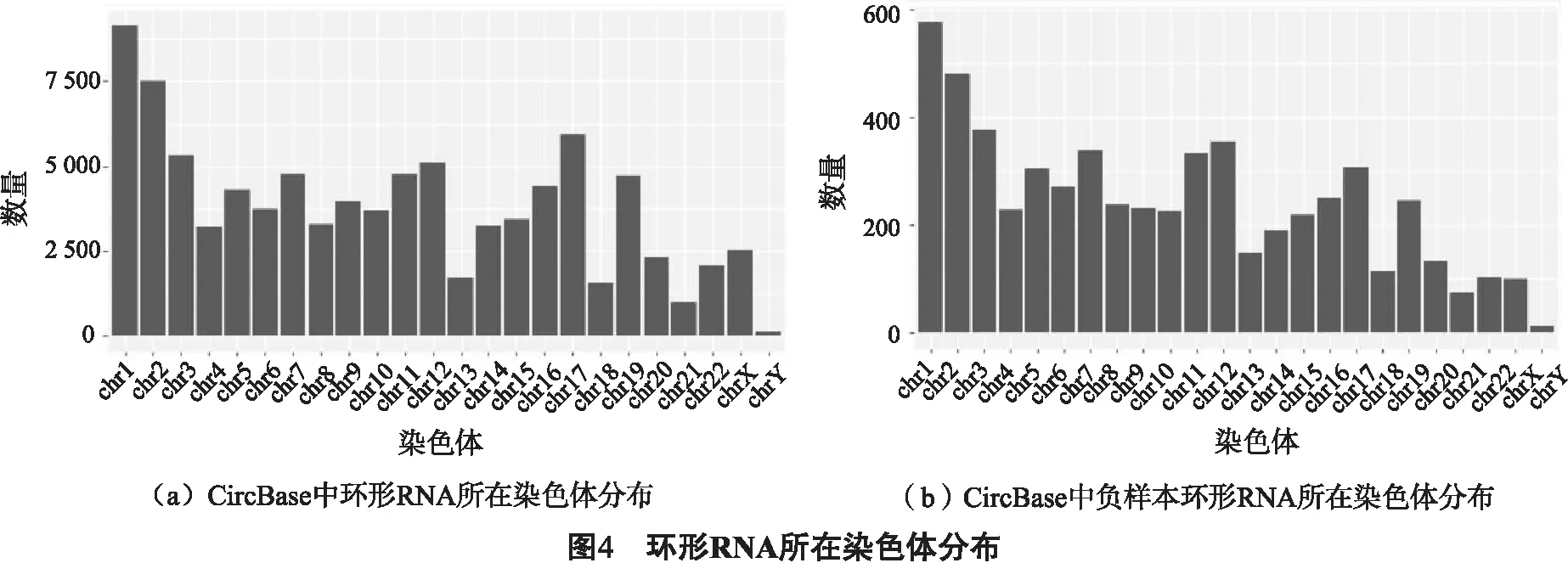
由于目前尚无确凿证据证明哪些环形RNA不能编码蛋白,但是已有研究统计,大约只有10%的环形RNA能够编码蛋白质.因此从CircBase数据库中随机筛选正样本之外的且与正样本等量的环形RNA作为负样本数据,共5 747个负样本.我们对随机筛选为负样本的环形RNA和数据库中所有的环形RNA的染色体来源进行了统计,结果如图4所示.我们发现2个数据集在染色体分布上基本一致,来源于1号染色体的环形RNA最多,来源于Y染色体的环形RNA最少,证明了我们随机挑选过程的科学性.
2.2 特征筛选
我们利用Python语言的sklearn库,分别构建了逻辑回归、支持向量机、随机森林和XGBoost分类模型.其中所用特征可大致分为7类,包括开放阅读框相关特征、内部核糖体进入位点相关特征、保守性相关特征、转录本编码蛋白相关特征、m6A修饰相关特征、特殊k-mer特征以及AUG含量和GC含量特征,详见表1.
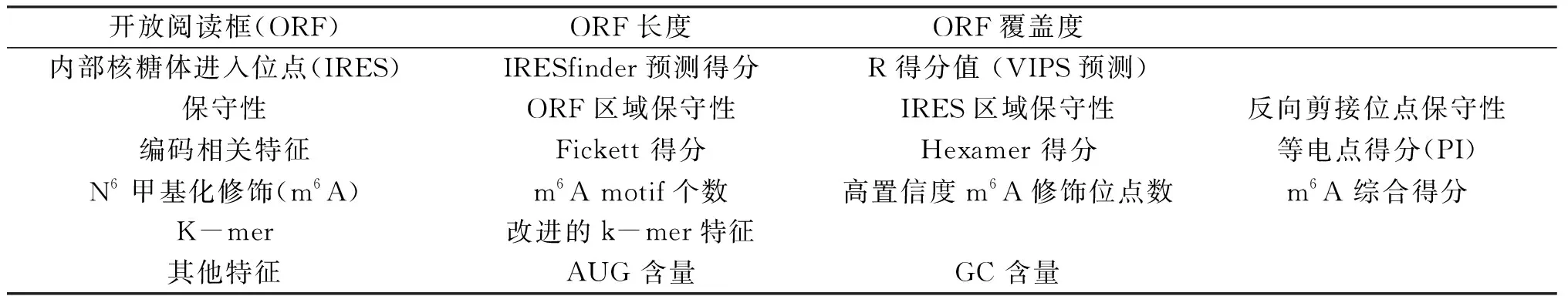
表1 环形RNA编码蛋白特征汇总
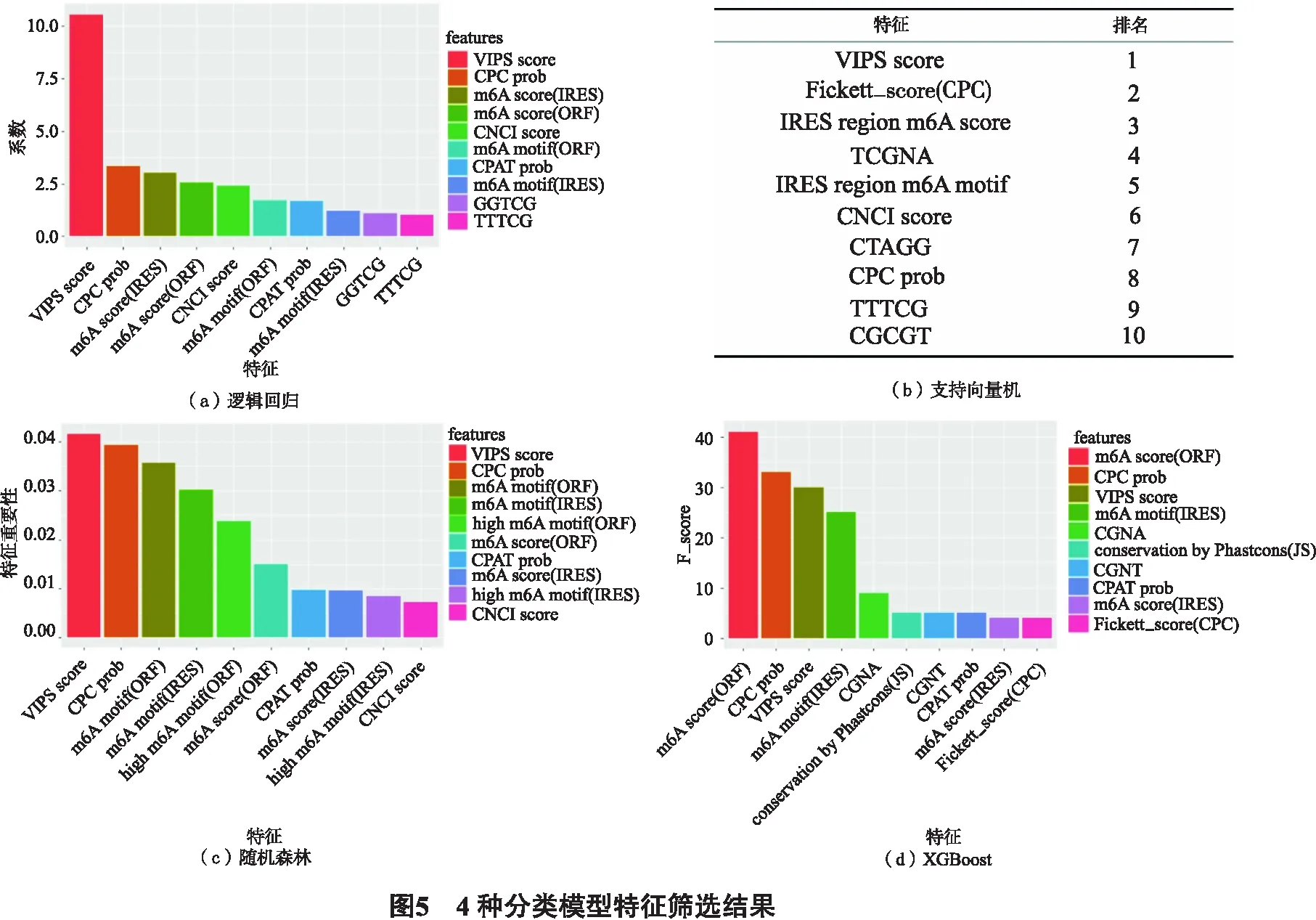
以上7类特征,共计2 525个特征,其中改进的k-mer特征2 500个.为防止模型的过拟合,对上述特征进行严格的特征筛选.对于逻辑回归模型,我们经过特征缩放,根据模型中各个特征系数大小的绝对值,取排名前10的特征作为训练模型所要保留的特征,结果如图5(a)所示.其中,VIPS预测的IRES得分值对于模型的贡献最高, m6A修饰相关特征对于模型分类性能也极为重要.
对于支持向量机模型,采用sklearn标准库中的RFE函数进行递归特征消除,反复迭代去除最不重要的特征.最终,筛选出对模型贡献最大的10种特征,如图5(b)所示.结果显示,VIPS预测的IRES得分值对模型贡献度排名最高,另外还有4个改进的k-mer特征对于支持向量机模型也很重要.
对于随机森林模型,使用feature_importances函数,对各个特征的重要性进行评估.从中选出最重要的10个特征,结果如图5(c)所示.同样的,VIPS预测的IRES得分值贡献度排名第一,m6A预测相关特征对于随机森林模型也很重要.
对于XGBoost模型,则通过计算各个特征的F-score值,判断特征的重要性.最终选出10个贡献度最大的特征,结果如图5(d)所示.排名前10的特征依次是开放阅读框区域m6A得分、CPC预测编码潜能得分、VIPS预测的IRES得分、IRES区域的m6A motif数量、CGNA(k-mer) 、Phastcons预测的环形RNA接头位置保守性得分、CGNT、CPAT预测的编码潜能得分、IRES区域的m6A得分和Fickett_score.
综合分析上述特征筛选的结果,VIPS预测的得分值在3个模型中贡献度最高,IRES区域的m6A修饰得分,m6A motif个数等参数对模型同样重要.上述结果表明,IRES元件以及m6A修饰对于环形RNA编码蛋白的调控机制尤为重要.
3 模型性能的评估
基于标准数据集,使用筛选后的最优特征,分别构建了逻辑回归、支持向量机、随机森林和XGBoost 4种分类模型.接下来对各个模型的分类性能进行评估,性能评估的方法分为10倍交叉验证和独立测试集验证2种方式.其中独立测试集是由文献验证的能够编码蛋白的环形RNA数据和等量的负样本组成.性能评估的指标包括预测准确率ACC、精确度PRE、ROC曲线下面积AUC等.
3.1 逻辑回归模型的性能评估
我们使用Python语言中的sklearn标准库计算逻辑回归模型分类结果的TP(真阳性值), TN(真阴性值), FN(假阴性值)和FP(假阳性值).将所有正负样本用作训练集,进行10倍交叉验证,受试者工作曲线(ROC曲线)如图6所示.
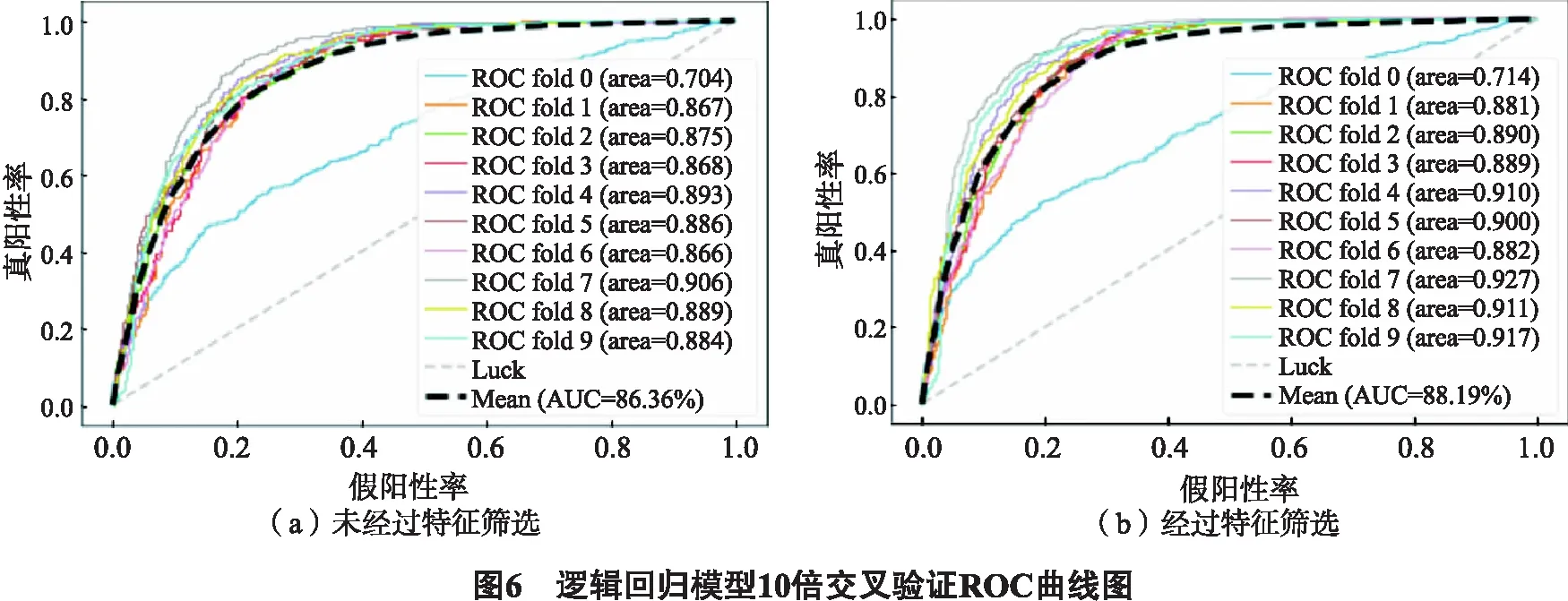
经过特征筛选,逻辑回归模型10倍交叉验证的平均AUC提升了1.83%,证明进行特征筛选防止模型过拟合的必要性.经过特征筛选后,10倍交叉验证的平均预测准确率达到了81.56%,精确度达到了78.34%.然而,在独立测试集上,逻辑回归模型的预测准确率仅有65%,效果并不理想.
3.2 支持向量机模型的性能评估
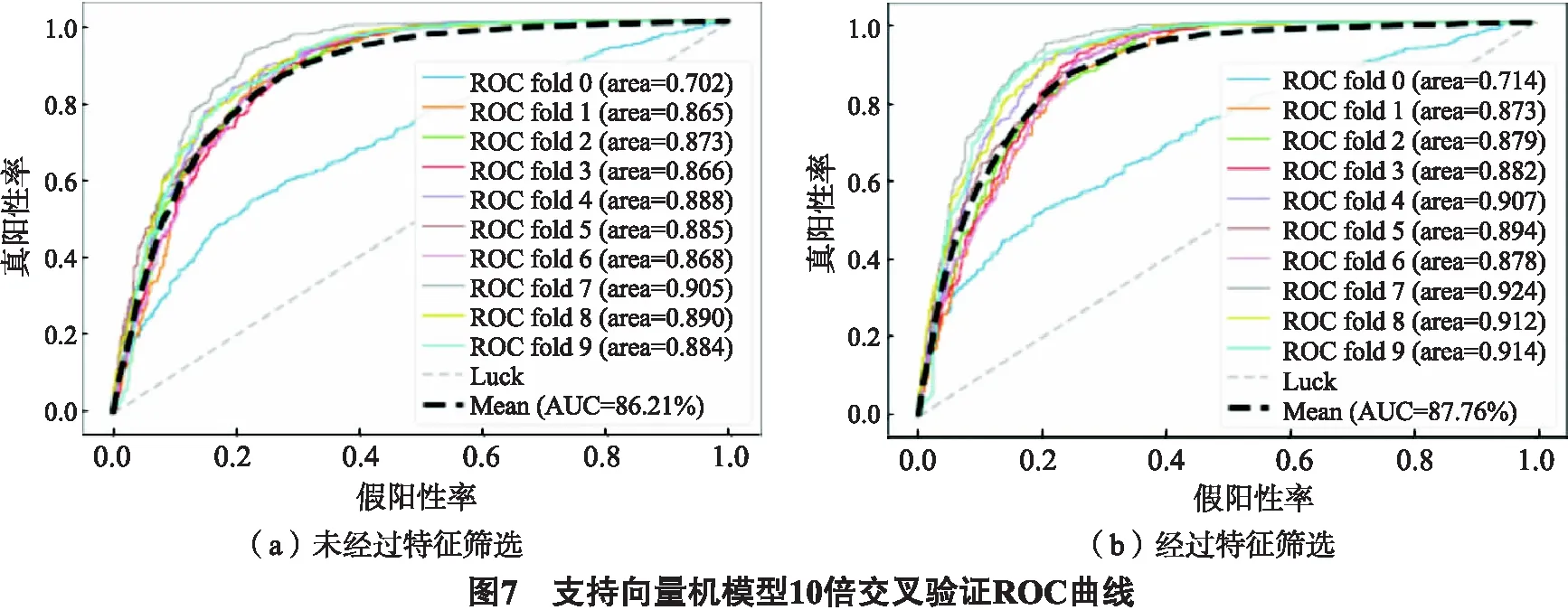
采用十倍交叉验证对高斯核(RF kernel)支持向量机模型进行性能评估,特征筛选前和筛选后的10倍交叉验证ROC曲线如图7所示.支持向量机模型经过特征筛选后平均AUC提升了1.55%,达到了87.76%;而10倍交叉验证的平均预测准确率达到80.71%,精确率达到75.78%.虽然10倍交叉验证的结果略低于逻辑回归模型,但在独立测试集上,支持向量机模型的预测准确率达到了70.6%,明显好于逻辑回归模型.
3.3 随机森林模型的性能评估
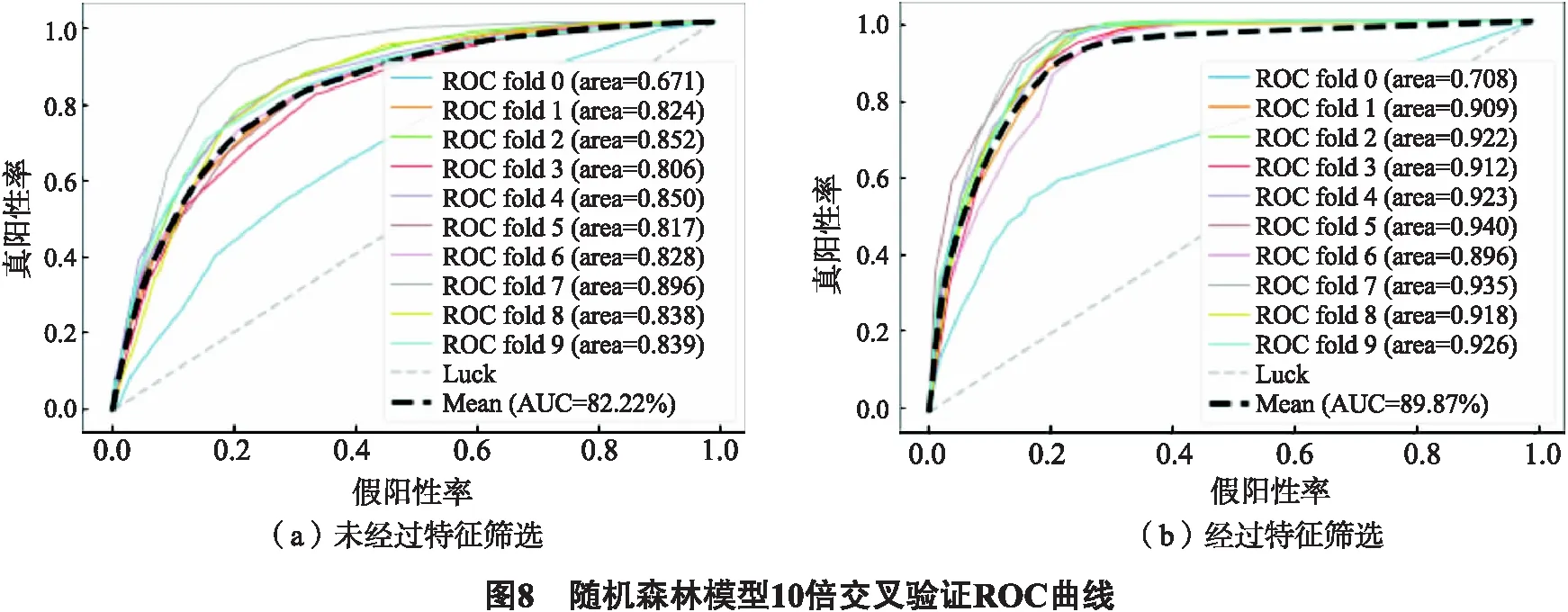
对于随机森林模型,首先使用所有特征构建分类模型,再对特征进行筛选,基于筛选出的特征再次构建模型.对先后构建的2个模型,分别进行10倍交叉验证,结果如图8所示.未经特征筛选的随机森林模型无论在AUC值上,还是在其它各类性能参数上均劣于逻辑回归模型和高斯核支持向量机模型.这可能是由于训练所用特征过多,构建的树节点过多导致模型过拟合,反而使其对真正有区分度的特征不敏感.经过特征筛选,随机森林模型10倍交叉验证性能提升明显,平均AUC提升了7.65%,达到了89.87%,证明了特征筛选过程的必要性.10倍交叉验证平均预测准确率达到了83.61%,精确度82.64%,略优于逻辑回归模型和支持向量机模型.独立测试集的测试结果显示,随机森林预测准确率为73.53%,优于逻辑回归和支持向量机.
3.4 XGBoost模型的性能评估
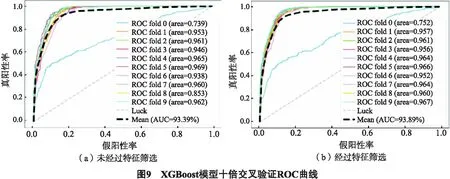
XGBoost模型是梯度提升算法的改进,应用于分类和回归问题.利用Python语言xgboost.sklearn包中的XGBClassifier函数构建分类模型.首先使用所有特征训练模型,接着筛选特征并根据筛选所得的特征训练模型,针对特征筛选前后的模型分别进行10倍交叉验证,结果如图9所示.XGBoost模型经过特征筛选,10倍交叉验证AUC值提升了0.6%,达到93%.XGBoost在10倍交叉验证上优于其他所有模型.经过特征筛选,10倍交叉验证的平均预测准确率为86.30%,精确度为80.98%,分类性能优异.在独立测试集上,XGBoost预测准确率为73.53%,与随机森林相同,优于逻辑回归和支持向量机模型.
3.5 小结与讨论
3.1-3.4部分先后采用逻辑回归、高斯核支持向量机、随机森林和XGBoost4种机器学习方法,使用筛选后的最优特征,分别在数据集上训练分类模型,并对其分类性能进行了评估.我们将各个模型的各项性能指标汇总如图10所示.
除了精确度略低于随机森林模型,XGBoost分类模型的各类性能指标均优于逻辑回归、支持向量机和随机森林模型.其10倍交叉验证的平均ROC曲线下面积达到了93.89%,分类准确率为86.30%,精确度指标为80.98%,分类性能明显优于其他模型.接着我们在独立测试集(来源文献验证的编码蛋白环形RNA)上对各类模型进行分类性能测试,性能评价汇总结果如图11所示,ROC曲线如图12所示.
在独立测试集上,支持向量机的预测结果偏向正样本,而随机森林的预测结果偏向负样本.为了进一步优化模型的分类性能,将XGBoost模型、随机森林模型和支持向量机模型融合,对环形RNA编码蛋白的潜能做出最终的综合评判,如图13所示.3个模型分别根据各自特征筛选的结果从总特征中提取对于模型贡献最大的10个特征,分别预测环形RNA是否能够编码蛋白,投票给出环形RNA编码蛋白潜能的预测结果.

接着对这个综合分类模型进行性能评估,分别进行10倍交叉验证和独立测试集上的验证,ROC曲线结果如图14所示.综合分类模型10倍交叉验证的平均AUC值达到了93.69%,只略微低于XGBoost10倍交叉验证的AUC值(逻辑回归88.19%、支持向量机87.76%、随机森林89.87%、XGBoost 93.89%),而其它各项性能指标均优于XGBoost模型,其中10倍交叉验证平均准确率提高了0.15%,达到了86.66%,精确度提高了0.88%,达到了82.07%.相比逻辑回归、支持向量机和随机森林模型,综合分类模型优势明显.在独立测试集上,综合分类模型预测准确率为73.53%,与XGBoost模型相同.
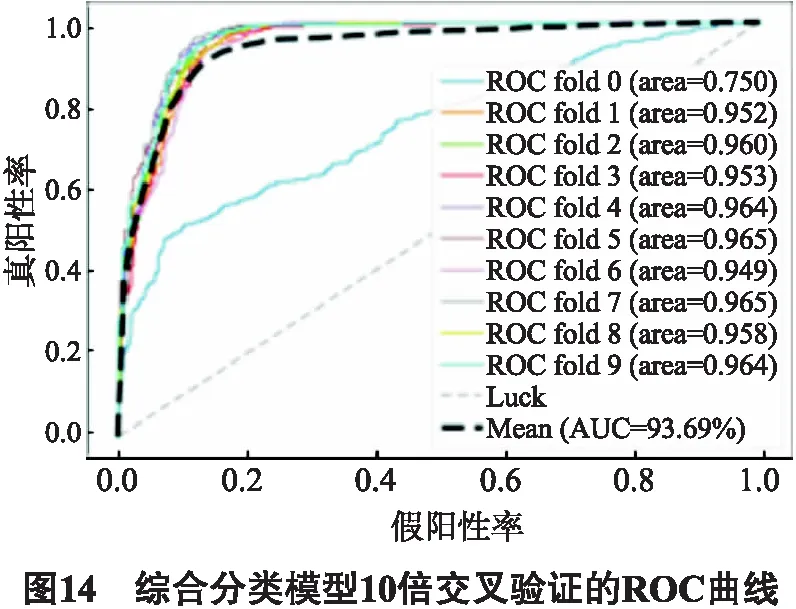
根据上述结果可以看出,综合分类模型相较XGBoost在分类性能上有所提升,与逻辑回归、支持向量机和随机森林方法相比具有较大的优势.据此,我们将综合分类模型用于构建最优的环形RNA编码蛋白潜能的预测分类器.
4 结语
本文首先分析了环形RNA的序列与结构特征,从ORF、IRES元件、m6A修饰等方面研究环形RNA编码蛋白的相关调控机制.其次,为预测环形RNA编码蛋白潜能,收集正负样本数据,分别构建了逻辑回归、支持向量机、随机森林和XGBoost分类模型.为避免模型的过拟合,对特征进行了严格筛选.评估了各个模型的分类性能,发现XGBoost分类模型性能明显优于其它模型.最后,构建由XGBoost、随机森林和支持向量机组成的综合分类模型,该模型优于任一单独预测模型.综合分类模型10倍交叉验证的平均AUC达到了93.69%,预测准确率达到了86.66%,能够较好的预测环形RNA编码蛋白的潜能.
本文首创性地基于环形RNA序列与结构特征,运用机器学习的方法预测环形RNA编码蛋白的潜能.现有的环形RNA编码蛋白预测工具(CircPro[19]、CircCode[20])都基于翻译组测序(Ribo-seq)数据,需要更高的实验条件,不够便利.然而,在独立测试集上,综合分类模型的预测准确率依然不高.随着越来越多的编码蛋白环形RNA被发现,高置信度训练集样本量的扩充可能对于模型的训练能够提供一定的帮助.当训练样本足够大时,还能将深度学习的方法应用于环形RNA编码蛋白的预测分类.此外,随着环形RNA编码蛋白机制的深入研究,相信会有更多更有效的编码蛋白环形RNA相关生物信息学工具以及数据库出现,反过来进一步促进编码蛋白环形RNA的发现及对其编码机制的研究.
