流式核酸单分子计数方法研究进展
2020-12-09王迪吴枭王志栋高运华
王迪,吴枭,王志栋,高运华
中国计量科学研究院前沿计量科学中心, 北京 100029
近年来,多种微量核酸样本的检测技术已经广泛应用于分子诊断、转基因标识、食品药品残留检测等各个领域[1]。尤其当前新冠病毒在全球肆虐[2-7],以实时荧光定量PCR(real-time quantitative polymerase chain reaction, real-time qPCR)为代表的核酸检测方法成为诊断的“金标准”,在抗击疫情中发挥了重要作用[8-10]。但同时,一些试剂盒灵敏度较低造成检测结果“假阴性”的报道屡见不鲜,给新冠肺炎的临床诊治造成了一定的困扰。因此研发更精确灵敏的检测技术,提升对微量核酸样本的定量测量能力,十分迫切。
流式核酸单分子计数是一种基于流式细胞术的核酸绝对定量方法,该方法无需PCR扩增直接检测单个核酸分子,通过计数的方式得到样本中核酸分子的拷贝数,从而直接溯源到国际单位制(international system of units, SI)。作为潜在的基准计量方法,它已经成为当前国际生物计量研究的热点,具有广阔的应用前景。本文在介绍流式核酸单分子计数方法原理的基础上,重点阐述了液流系统、光电系统和数据分析模型的研究发展历程,并展望了该方法的发展前景。
1 流式核酸单分子计数的基本原理
流式核酸单分子计数的原理与传统的流式细胞术相似,它最初的检测装置就是通过改造流式细胞仪研制的。两者均是在驱动荧光标记样本的过程中,以直线状态依次逐个流经检测区并被激光激发产生荧光信号,通过采集信号对样本进行鉴定分析[11-13]。两者的主要区别在于:①前者检测对象为核酸单分子,后者为单细胞。由于核酸分子是细胞大小的几千分之一,因此前者对灵敏度的要求非常高。②前者侧重定量测量,后者侧重定性鉴定[14]。
流式核酸单分子计数方法以荧光标记的核酸单分子为研究对象,主要采用了微流控系统和激光诱导的荧光(laser induced fluorescence, LIF)检测技术。通过压力或电场的作用使极限稀释的核酸样本在流道中实现流体聚焦——以单分子状态在微纳米级通道中流动,并在经过检测区时逐个被激光激发产生荧光信号。这些荧光被特定波长的滤光片过滤后通过光电元件进行光学成像和光电转换放大,然后由电脑的分析软件识别信号并读取核酸分子的拷贝数,并通过数学模型运算得到最终定量结果,从而实现对样本的绝对定量。
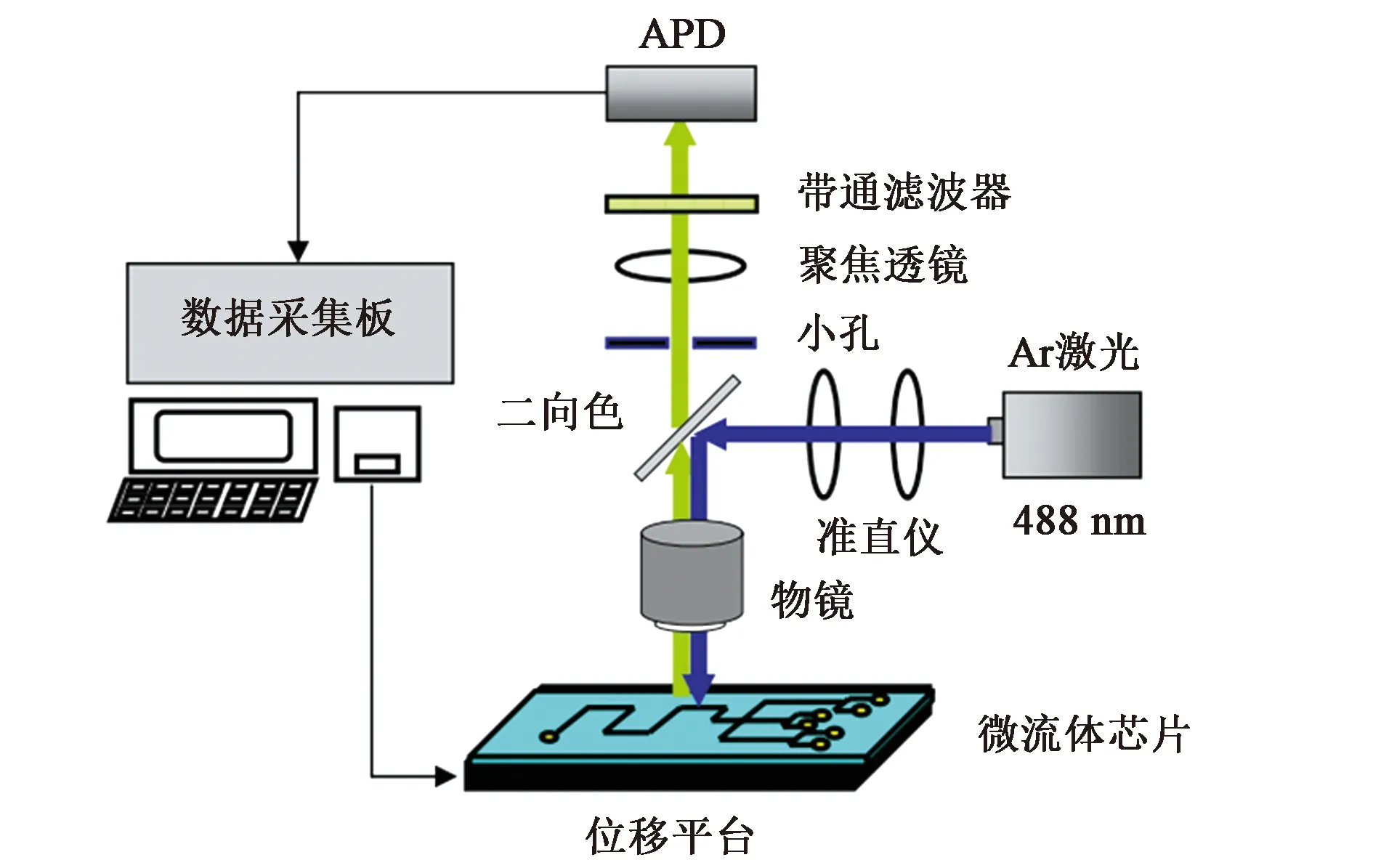
图1 流式核酸单分子计数测量装置[14]Fig.1 Measuring equipment of flow cytometry counting for single nucleotide molecule
2 几种主要核酸计量方法的比较
目前用于核酸分子定量的主要技术包括:紫外分光光度法、荧光染料法、电感耦合等离子体-发射光谱(inductively coupled plasma-emission spectrometry, ICP-OES)、同位素稀释质谱(isotopic dilution mass spectrometry, IDMS)、高分辨率电感耦合等离子体质谱(high resolution-inductively coupled plasma-mass spectrometry, HR-ICP-MS)、实时荧光定量PCR和数字PCR法(digital PCR, dPCR)等。
采用紫外分光光度法测定核酸含量简便快捷,但灵敏度较低且易受溶液pH、苯酚、蛋白质、多糖等的影响[15-17]。荧光染料法通过SYBR Green I或PicoGreen等荧光染料标记双链DNA,根据激发产生的荧光信号强度定量DNA浓度。荧光染料法具有很高的灵敏度,但定量结果易受到DNA纯度、DNA减色效应的影响,且测量痕量DNA样本时偏差较大,准确性和重复性不佳[18]。ICP-OES主要基于DNA中磷元素含量的测定对DNA浓度进行定量,该方法的扩展不确定度较小,定量结果可溯源至SI单位(摩尔)。但ICP-OES定量的前提是有效的去除其他物质中磷的干扰,并定量消解过程磷的回收率;DNA样品量较大(mg级)且具有足够高的纯度,这限制了该方法的实际应用[19-21]。IDMS是一种将分析物的同位素标记物作为定量内标的质谱定量技术,具有很高的定量精密度,是目前国际物质量咨询委员会(Consultative Committee on the Quantity of Material,CCQM)认可的潜在的基准方法。2019年,美国国家标准与技术研究院建立了酸解同位素稀释质谱法,利用甲酸水解核酸片段实现对RNA、短的线性DNA以及基因组DNA的准确定量[22]。但总体来讲,该方法对DNA纯度要求也很高,且前期的核酸酶解或酸解必须彻底[21]。HR-ICP-MS法通过磁场和电场来分选离子,质量分辨率高,量值溯源清楚,定量准确。灵敏度可达到pg级,但仍无法满足痕量DNA样本的检测要求[23-26]。
数字PCR是一种近十多年来快速发展起来的、基于核酸单分子计数的绝对定量方法。它将低浓度的核酸样本平均分配到大量的独立反应单元中进行扩增反应,根据泊松分布和阳性信号比例来定量特定核酸片段的拷贝数[27-28]。相比real-time qPCR,它具备一些明显的优势,如灵敏度更高,对PCR抑制剂不敏感,无需借助标准品即可对核酸浓度绝对定量,定量结果不受扩增效率影响等[29-31]。随着数字PCR仪的商品化,dPCR技术已越发成熟,经常被用于核酸标准物质的定值研究以及核酸样本绝对定量的国际比对,成为核酸计量研究的重要技术[32-35]。
与上述技术相比,流式核酸单分子计数具有以下显著特点:①直接检测单个核酸分子,无需PCR扩增,避免了环境和检测试剂的污染对检测结果造成影响;②通过计数的方式直接读取样本的拷贝数,与数字PCR一样无需核酸外标,避免了标准品与实际样本检测效率差异带来的定量误差[36-37]。因此,研究建立流式核酸单分子计数方法,是对现有核酸计量方法的重要补充,将显著提升对微量核酸样本的精准定量测量能力,并进一步推动我国核酸量值溯源传递体系的发展。
3 流式核酸单分子计数检测体系的研究发展历史
流式核酸单分子计数不需要PCR扩增,直接通过计数的方法对核酸分子定量。建立该方法需要解决两个核心问题:①单个核酸分子不经扩增结合的荧光染料很有限,必须尽可能增强激发效率,显著提升检测的灵敏度才能识别单分子的荧光信号;②核酸分子非常小,保证在激光激发的检测区内同一时间只通过一个分子是拷贝数定量的关键。
为解决上述问题,流式核酸单分子计数检测体系在传统流式基础上进行了一系列的优化改造。下面将具体介绍其三个组成部分——液流系统、光电系统和数据分析系统的研究发展历史,并由此探究该方法的具体定量策略。
3.1 液流系统
搭建适用的液流系统,使标记后的核酸分子依次逐个经过检测区被激光激发,即保证在激光束与样本流正交重合的检测体积(probe volume, PV)内,同一时间除个别冲突事件和DNA二聚体外最多只有一个核酸分子通过(这与数字PCR每个反应单元中最多含有一个DNA分子相似)。因为核酸单分子只有纳米级大小,若实现在一个PV内只有一个核酸分子,必须满足两个条件:①核酸浓度应足够低,一般102~104拷贝·μL-1;②PV足够小(pL级),即要求样本流和激光束高度聚焦,样本流一般控制在水平方向20 μm以内。同时这对促进激光与核酸分子正交也非常重要,是提高检测灵敏度和减少测量误差的关键。
1993年,美国Los Alamos国家实验室首次建立了超灵敏流式细胞仪定性鉴定不同大小DNA片段的方法。Goodwin等[38]采用内径250 μm的流动池,通过重力驱动鞘液流和DNA样本流,使鞘液流速约30 μL·min-1,仅为传统流式流速的几百分之一。在鞘液压力的作用下样本流聚焦在直径20 μm的范围内,样本流速小于1 μL·min-1。他们发现λ-DNA酶切片段用染料TOTO-1标记后,应用该系统可根据荧光信号强度的差异来区分10.0~48.5 kb不同长度的DNA片段(DNA片段的荧光强度与片段大小成正比),鉴定效果显著优于传统电泳的方法。
为降低样本通过检测区时的流速,随后Petty等[39]又在流动池出口处填加一个内径280 μm的PE管以增加液流阻力,从而延长了单个DNA分子通过检测区时的曝光时间。改造后该系统可有效识别1.5 kb的核酸片段。1999年Yan等[40]以石英毛细管作为样本流道,进一步缩小了样本流宽度(16 μm)和激光检测体积(约9 pL);并且比较PicoGreen、TOYO-1和SYTO 16等9种常用的核酸荧光染料标记λ-DNA酶切片段后的检测效率,发现PicoGreen标记产生的荧光信号不仅信噪比高,且核酸片段大小与荧光信号强度的线性关系不会因其浓度的变化而明显改变。
2003年,Zheng[41]的工作显著推动了该技术在核酸绝对定量方面的应用。他们采用毛细管电泳的方法实现流体聚焦,即在石英毛细管两端加上电极,当样本被压力驱动进入石英毛细管后,通过电场将DNA分子聚焦在液流中心约10 μm的区域内。从而进一步提高了激光对核酸分子的激发效率,并减少了测量误差。应用他的方法,可准确定量浓度在10~1 000 amol·L-1的λ-DNA片段。
韩国标准科学研究院(Korea Research Institute of Standards and Science, CRISS)正式将流式核酸单分子计数方法应用于核酸计量领域。Lim等[37]参照上述毛细管电泳的方法搭建了一套测量装置,对λ-DNA分子定量的重复性误差小于3%。Yoo等[42]进一步升级该系统,并优化了电压和液流压力等测量条件,成功实现了对4.3 kb的pBR322质粒的定量测量,2016年该团队采用该方法参加了pBR322浓度的国际比对,测定的浓度与dPCR和IDMS的结果基本一致[43]。
3.2 光电系统
相比液流系统,流式核酸单分子计数装置的光电系统变化较小。Goodwin等[38]采用的光电系统设计如下:低功率的Ar+激光器设置在与液流管道90°垂直的位置,它发出的激光由一系列透镜聚焦后在样本流中央处,形成中心直径为40 μm的球形光斑。激发DNA分子产生的荧光信号被40×0.85 NA的物镜收集后,通过光栅和滤光片扣除非特异的光信号。然后将特定波长的荧光传输给光电倍增管(photomultiplier tubes, PMT)进行光电转换和信号放大,最后使用脉冲计数器进行原始计数并将数据传输给电脑进行数据分析。
随后的相关工作主要基于上述设计,通过不断优化光路和升级光电元件,来提高信噪比和检测灵敏度。比如采用外壁更薄的石英流动池或盖玻片,以适应物镜的工作距离,增强镜头的分辨率[39, 44]。或将激光聚焦更充分形成仅20 μm的光斑,采用60×的镜头收集荧光信号[42]。还有的研究在带通滤光片前设置一个50 μm的针孔以降低背景噪音[14]。
此外不同实验室采集处理荧光信号的方式略有不同。比如Zheng等[41]采用增强型电荷耦合元件(charge coupled device,CCD)进行高频率的荧光成像来捕获荧光信号,然后使用Image J软件对图片进行数据分析。而Yoo等[42]在荧光信号的接收端设置了电荷耦合元件(CCD)和雪崩光电二极管(avalanche photodiode, APD)双系统,即通过APD收集荧光信号并对单光子计数;通过CCD成像实时检测液流流动状态,并由此优化检测条件。
3.3 数据分析系统
对核酸浓度准确定量,必须开发出科学的数据分析系统,包括建立核酸单分子识别模式,以及修正原始计数结果的数学模型。
其中高效识别核酸单分子是数据分析的基础,具体的策略是:在直方图中可将核酸单分子产生的数字脉冲信号拟合成一个高斯分布曲线。把得到的原始数据去除背景噪音后,根据脉冲信号波谷处的信号大小设置阈值。若在特定时间内(bin)某一脉冲信号强度均高于设定的阈值,则对其计数。上述数据处理过程是通过研究人员自己研发的软件来实现的[29,38,40]。Ferris等[45]分析了单分子检测的统计结果,通过比较变异系数(coefficient of variation, CV)、不同浓度与计数结果的线性关系一步验证了该单分子识别模式的有效性。
另外,通过数学模型对原始计数结果进行校正是另一个关键问题。如修正APD光子响应间隔时间的影响、解决双粘体和偶发事件的干扰、清除非特异背景噪音等。
Habbersett等[46]发现APD在识别相邻光子间存在一定时间间隔(dead time, DT),即当DNA分子产生高密度光量子时,在该段间隔时间产生的信号不会被APD采集,从而造成计数偏低。Hussels等[36]提出可根据公式(1)对计数结果进行校正。其中N0指经DT优化后的计数结果,Nr指DT优化前的计数结果,τ为平均波峰宽度,T为总的计数时间。
(1)
另一方面,实际检测时样本中少量DNA分子会不可避免的形成二聚体,或发生冲突事件——即两个分子相距很近被当成一个分子计数,造成定量结果偏低。研究人员发现通过检测区时,若两个DNA分子形成二聚体或同时通过,则信号峰的高度(最大荧光信号强度)是单分子峰高度的近2倍;若双分子间距很小造成两个峰连在一起,则总峰宽(通过检测区的时间)是单分子峰的近两倍;若两个分子间距介于上述两种情况之间,则峰宽和峰高是单分子的1~2倍,峰面积(经过检测区产生的信号之和)是单分子的近2倍。因此比较峰的面积、高度和宽度,就可以鉴别出二聚体和冲突事件,对计数结果进行修正。
此外为清除非特异信号的影响,需设置只含有荧光染料的空白对照管,在样本的计数结果中减去空白对照的计数值(Ndye)。再根据测量样本得到的直方图中峰高大小来鉴定噪音信号(Nnoise),将其从总计数值中扣除。因此最后修正的核酸浓度Cc是由公式(2)计算得到,其中V指核酸样本体积,N0同公式(1),Ndw和Ndh分别表示通过峰宽和峰高鉴定的二聚体或冲突事件的计数值[42]。
Cc=(N0+Ndw+Ndh-Ndye-Nnoise)/V
(2)
4 展望
本文综述了流式核酸单分子计数绝对定量的原理,以及其液流系统、光电系统和数据分析系统的研究进展。在该方法的发展过程中,美国Los Alamos 国家实验室建立了基于流式的核酸单分子检测方法。他们通过分离不同大小的单个DNA片段进行微生物的物种定性鉴定,为进一步的定量测量奠定了扎实的基础。2016年DNA拷贝数绝对定量的国际比对(CCQM P154)中,流式核酸单分子计数与数字PCR两种方法对pBR322质粒的定量结果基本一致[43]。这表明流式核酸单分子计数作为一种潜在的核酸计量方法,将在生物计量领域发挥越来越重要的作用。
但是尽管流式核酸单分子计数已经成为当前国际生物计量领域的研究热点,目前对微量核酸样本的绝对定量仍主要以数字PCR为主。依托商品化的检测装置,数字PCR技术已较成熟,而流式核酸单分子计数的检测体系仍需不断完善,以进一步提升测量的稳定性和准确性。
此外该方法尚存在的一个主要技术问题亟需解决,即靶向核酸片段定量。目前该方法采用荧光染料非特异结合DNA双链,无法有效区分不同的核酸片段,进而对特异序列进行定量测量。因此只有当样本中含有的一种DNA片段或几种DNA片段间大小差异较大时才可采用该方法,这严重限制了它的应用范围。研究建立该方法的靶向标记技术,通过标记特异性序列的荧光基团实现对特定核酸片段的定性鉴定和定量测量非常重要,这直接决定了该方法能否应用于实际的分子检测工作,发挥其检测速度快、无PCR污染、定值结果直接溯源到SI单位等优点。
综上所述,作为世界核酸计量研究的热点,流式核酸单分子计数是一种潜在的核酸计量基准方法。建立和发展该方法,将显著提升对微量样本的精准定量测量能力,推动生物计量标准体系的构建。
