原子荧光光谱法测定土壤和沉积物中铋
2020-11-24周资凯鲍益帆叶昌林
周资凯,鲍益帆,叶昌林
(1.南昌大学资源环境与化工学院,南昌 330031; 2.台州市台环环境检测科技有限公司,浙江台州 318020)
土壤是环境的重要组分,由各种复杂物质构成,其中人类现代化活动所排放的污染元素长期积累于环境土壤中。铋为稀有元素,以游离金属和矿物形式存在,广泛应用于半导体产业、涂料工业、医疗等领域[1],现行国家标准GB 15618–2018 《土壤环境质量 农用地土壤污染风险管控标准(试行)》和GB 36600–2018 《土壤环境质量 建设用地土壤污染风险管控标准(试行)》均未规定铋的限值[2–3]。铋非人体必需元素,具有细胞毒性和弱放射性,可累积于人体肾脏,引起慢性中毒。环境土壤中的铋元素,容易通过食物链或其它活动进入人体,进而影响人体健康,因此准确测定土壤中铋元素含量具有重要意义。
原子荧光光谱法测定元素含量具有基体干扰少、灵敏度高、检出限低、仪器分析成本低等特点,广泛应用于痕量和超痕量元素分析,是目前测定土壤和沉积物中铋元素的常用方法。土壤和沉积物样品前处理消解方法主要有水浴法[4–6]、电热板法[7–8]和微波法[9–12]。水浴法加热温度平稳、恒定,区域受热均匀,但加热缓慢、费时;石墨加热板法易引起瞬间加热脉冲,且局部温差较大;微波法以高氯酸为消解液,对于含有有机物的样品,在高温高压下易引起爆炸,存在一定的危险。
铋属于易挥发元素,高温环境下易造成挥发损失,因此在样品前处理和分析中需要严格控制操作过程,否则会对测定结果造成一定影响;同时由于土壤成分基体复杂,存在多种干扰,采用HJ 680–2013 《土壤和沉积物 汞、砷、硒、铋、锑的测定 微波消解/原子荧光法》测定时,发现同一样品测定结果的相对标准偏差相差较大,精密度不高,难以满足测定要求[13]。笔者以国家一级土壤成分分析标准物质为研究对象,通过酸组分优化和试验条件改进,对3 种消解方法进行综合效益对比,确定了最佳消解方法和样品处理条件。该方法于同一消解管中可完成样品消解、定容和上机测定[14],避免了多步骤转移带来的测定误差,提高了分析结果的准确度。该方法已应用于环境土壤和沉积物样品中铋元素的测定,为高通量样品中痕量铋的检测提供了简便、快速和准确可靠的方法。
1 实验部分
1.1 主要仪器与试剂
原子荧光光度计:PF31 型,北京普析通用仪器有限责任公司;
电热板加热器:S36 型,北京莱伯泰克仪器股份有限公司;
电热恒温水浴锅:DK–98–22A 型,天津市泰斯特仪器有限公司;
多通量微波消解仪:Jupiter–B 型,上海新仪微波化学科技有限公司;
电热鼓风干燥箱:WGLL–230BE 型,天津市泰斯特仪器有限公司;
分析天平:CPA225D 型,感量为0.01 mg,赛多利斯科学仪器(北京)有限公司;
盐酸:优级纯,含量为36%~38%,浙江汉诺化工科技有限公司;
硝酸:优级纯,含量为65%~68%,上海阿拉丁生化科技股份有限公司;
硫酸:优级纯,含量为95%~98%,国药集团化学试剂有限公司;
氢氟酸:分析纯,含量不小于40%,上海凌峰化学试剂有限公司;
过氧化氢:分析纯,含量不小于30%,上海凌峰化学试剂有限公司;
高氯酸:分析纯,含量为70%~72%,上海阿拉丁生化科技股份有限公司;
国家一级土壤成分分析标准物质:GBW 07430[GSS–16,珠江三角洲,铋含量为(1.44±0.11) mg/kg],GBW 07446 [GSS–17,内蒙古乌拉特后旗沙化土,铋含量为(0.15±0.02) mg/kg],GBW 07452[GSS–23,浙江省象山东海滩涂沉积物,铋含量为(0.44±0.03) mg/kg],中国地质科学院地球物理地球化学勘查研究所;
实验用水为桶装娃哈哈饮用水,杭州娃哈哈集团有限公司;
所有实验用玻璃器皿、消解管使用前皆以硝酸溶液(1∶1)浸泡24 h,洗净、晾干备用;混合酸消解液为预混合,所有药剂溶液均现用现配。
1.2 仪器工作条件
原子器温度:200oC;灯源:铋灯源;灯电流:40 mA;原子化器高度:8 mm;光电倍增管负高压:280 V;载气:氩气,流量为300 mL/min;屏蔽气:氩气,流量为600 mL/min;分析读数时间:25 s;延迟时间:4 s;环境温度:(25±5)oC,环境湿度:(55±5)%。
1.3 样品采集与制备
依 据GB 17378.3–2007[15],HJ/T 166–2004[16]和NY/T 395–2012[17]相关规定采集、运输、制备及保存样品(农业、建筑用土和管道淤泥沉积物样品),避免过程污染和损失。样品于实验室中进行挑拣(去除杂物),然后分成2 份,一份摊平阴干,阴干后以对角线四分法手工捣碎,过2.0 mm(10 目)筛,封存,待测;另一份在原始含水状态下进行测定。均以HJ 613–2011[18]法对样品进行干物质和水分含量测定。
1.4 样品处理及测定
取制备好的土壤和沉积物样品或国家一级土壤成分分析标准物质(0.100 0±0.000 5) g 置于25 mL 玻璃消解管中,加入0.5~1.0 mL 水润湿,再加入盐酸–硝酸–硫酸消解液,然后将消解管置于100℃水浴锅中加热消解0.5~1 h,全程加盖并以纱布和皮筋扎紧(每隔15 min 晃动一次),消解完成后,以水定容至标线,摇匀待测。
土壤样品中铋元素含量按式(1)计算:
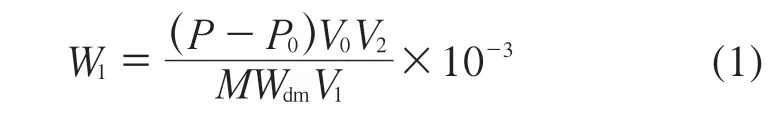
式中:W1——土壤样品中铋元素含量,mg/kg;
P——标准工作曲线查得铋的浓度,μg/L;
P0——空白式样中铋的浓度,μg/L;
V0——定容体积,mL;
V1——分取体积,mL;
V2——分取后测定试液的定容体积,mL;
M——土壤样品质量,g;
Wdm——土壤样品中干物质含量,%。
沉积物样品中铋元素含量按式(2)计算:
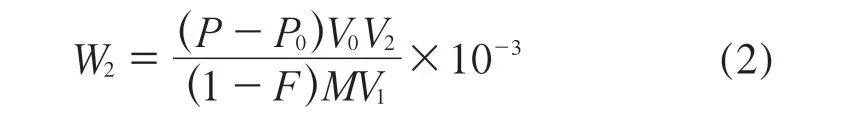
式中:W2——沉积物样品中铋元素含量,mg/kg;
P——标准工作曲线查得铋的浓度,μg/L;
P0——空白式样中铋的浓度,μg/L;
V0——定容体积,mL;
V1——分取体积,mL;
V2——分取后测定试液的定容体积,mL;
M——沉积物样品质量,g;
F——沉积物样品含水率,%。
2 结果和讨论
2.1 消解液选择
将盐酸、硝酸、硫酸、氢氟酸、高氯酸和过氧化氢[12,19]搭配组合,共组成13 种消解体系,在消解时间均为1 h 条件下,分别考察不同消解体系对土壤成分分析标准物质测定结果的影响,结果见表1。
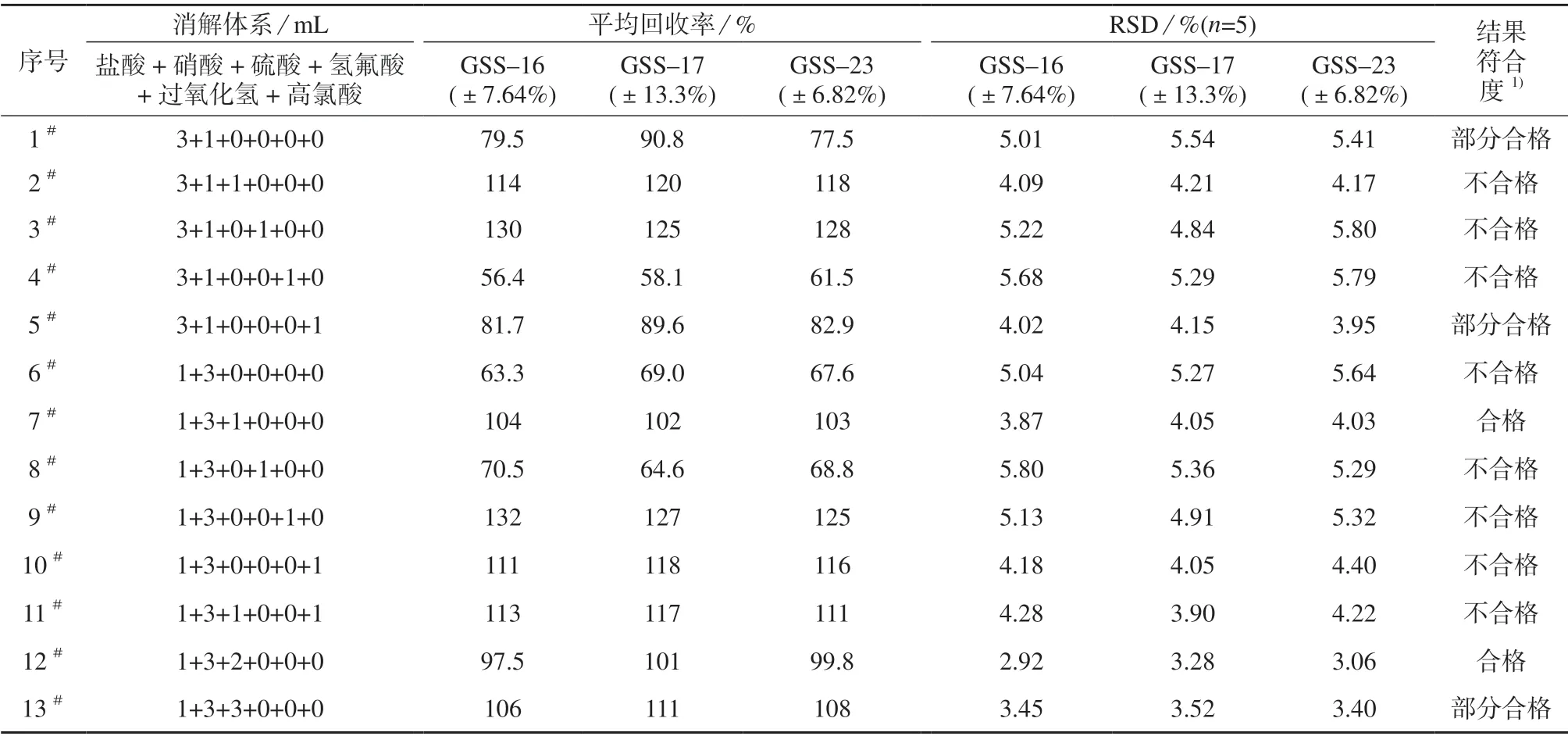
表1 不同消解体系试验结果
由表1 可知,1#~10#中仅7#消解体系的测定结果符合度合格,加入硫酸和高氯酸的消解体系(2#,5#,7#,10#)的精密度较为稳定。将7#进一步优化为11#~13#,发现12#与7#消解体系的测定结果均有较高的准确度,而12#的精密度更好。综合考虑,采用盐酸–硝酸–硫酸(体积比为1∶3∶2)三酸体系作为消解液。
2.2 消解方法选择
以盐酸–硝酸–硫酸(体积比为1∶3∶2)三酸体系为消解液,分别采用水浴法、电热板法和微波法对土壤成分分析标准物质GSS–23 进行消解,在1.2仪器工作条件下测定,结果见表2。由表2 可知,水浴法在100oC 下分别消解0.5,1 h 时,测定结果的相对标准偏差分别为3.27%和3.06%,平均回收率分别为97.5%和99.8%,表明该方法回收率符合要求,且精密度较高;电热板法则需消解6 h 以上回收率才满足要求,且精密度偏低;微波法3 组消解条件下回收率均满足要求,但精密度皆相对偏低。
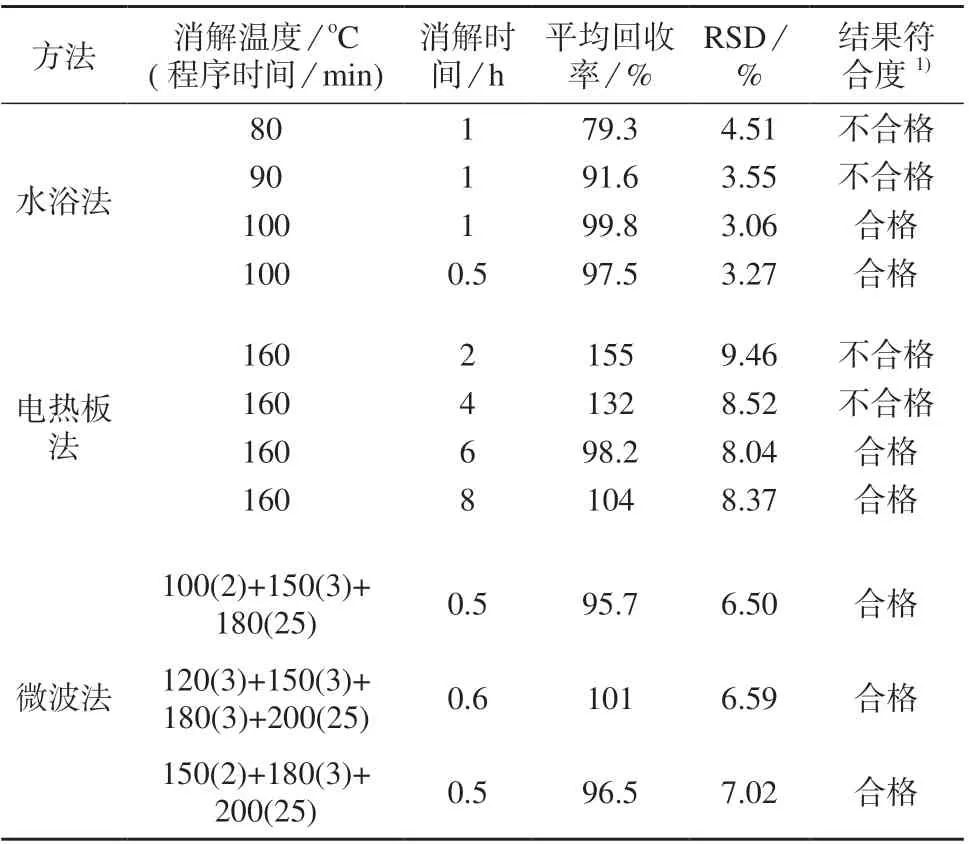
表2 不同消解方法试验结果(n=5)
不同消解方法结果对比见表3。由表3 可知,电热板法总耗时较长,为7.5 h;微波法和水浴法消解时间较短,均为0.5 h,但微波法后续赶酸增加了额外处理时间,总耗时为7.0 h。3 种方法的回收率均满足测定要求,与水浴法相比,电热板法和微波法的精密度偏低。综合考虑,选择水浴法在100oC 下对样品进行消解0.5 h。
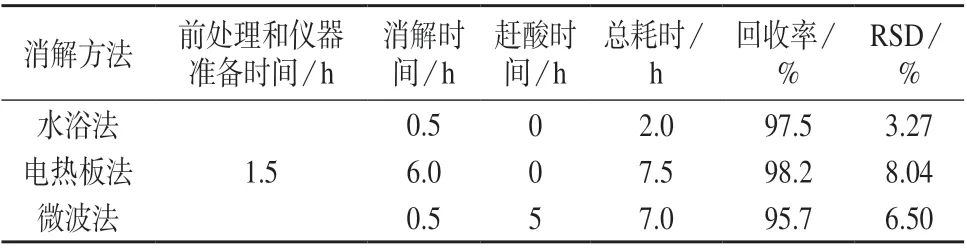
表3 不同消解方法结果对比(n=5)
2.3 样品含水量对测定结果的影响
为考察样品含水量对铋元素测定结果的影响,将采集的土壤样品分为两部分,一部分直接测定,另一部分风干后测定。土壤标准物质则分为加水和不加水两种情况测定。标记S 代号的为加水模拟含水样品(加水量大于5.00%),标记X 代号的为样品加水后再风干样品,测定结果见表4。由表4 可知,标记S 代号的加水模拟样品测定结果均降低,原因是水分含量增加,稀释了酸组分,降低了对样品的消解能力,且含水量增加导致消解过程中蒸汽量增大,影响消解管闭合程度,此外在消解完开盖定量时,少量铋元素随蒸汽流失,导致测定结果偏低[20];标记X代号的再风干样品测定结果亦有所降低。由此可见,样品含水量较大,会导致测定结果偏低,且样品处理环节越多、步骤越复杂,将会影响测定结果准确度。

表4 样品含水量对分析结果的影响(n=5)
2.4 线性方程与检出限
在1.2 仪器工作条件下,仪器自动稀释铋元素质量浓度为0.0,0.1,1,2,4,6,8,10 μg/L 进行测定,以铋的质量浓度(x)为横坐标、荧光强度(y)为纵坐标进行线性回归,计算线性方程和线性相关系数。结果表明,铋的质量浓度在0.0~10.0 μg/L 范围内与荧光强度线性关系良好,线性方程为y=109.31x–7.860,相关系数为0.999 9。
按1.4 方法平行制备11 份空白样品,在1.2 仪器工作条件下进行测定,计算11 次测定结果的标准偏差,以3 倍标准偏差对应的浓度作为方法检出限,得检出限为0.01 μg/L,将3 倍检出限定义为测定下限。经1.4 中式(1)和(2)换算,铋的检出限为0.008 mg/kg,测定下限为0.024 mg/kg。
2.5 精密度试验
选取国家一级标准物质GSS–16,GSS–17 和GSS–23,分别平行称取12 份,按1.4 方法进行样品处理,在1.2 仪器工作条件下测定,结果见表5。由表5 可知,测定结果的相对标准偏差为2.07%~3.84 %,表明该方法精密度良好,满足测定要求。
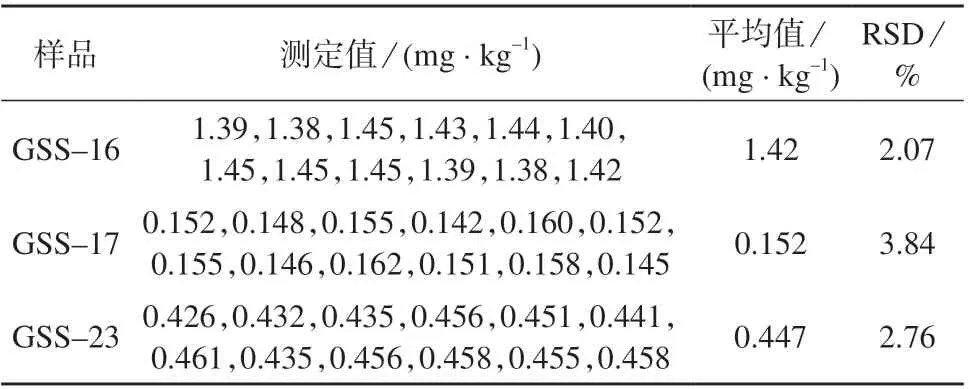
表5 精密度试验结果
2.6 准确度试验
2.6.1 标准样品测定
准确称取国家一级土壤标准物质GSS–16,GSS–17,GSS–23,按1.4 方法进行样品处理,在1.2仪器工作条件下测定,结果见表6。由表6 可知,3种标准样品的测定值均在标准值不确定度范围内,表明该方法准确度较高。
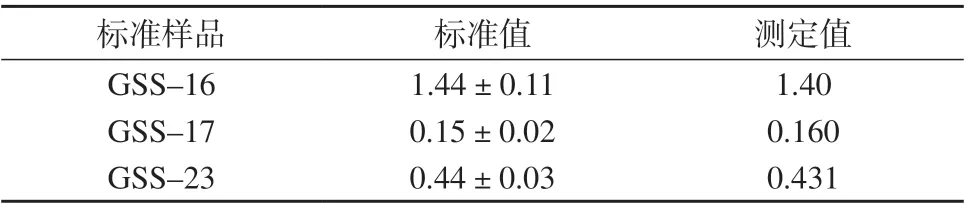
表6 标准样品测定结果 mg/kg
2.6.2 加标回收试验
准确称取农业用土、建筑用土和管道淤泥沉积物样品各3 份,分别加入不同浓度的铋标准溶液,按1.4 方法进行样品处理,在1.2 仪器工作条件下测定,结果见表7。由表7 可知,样品加标回收率为101%~119%,满足分析要求。
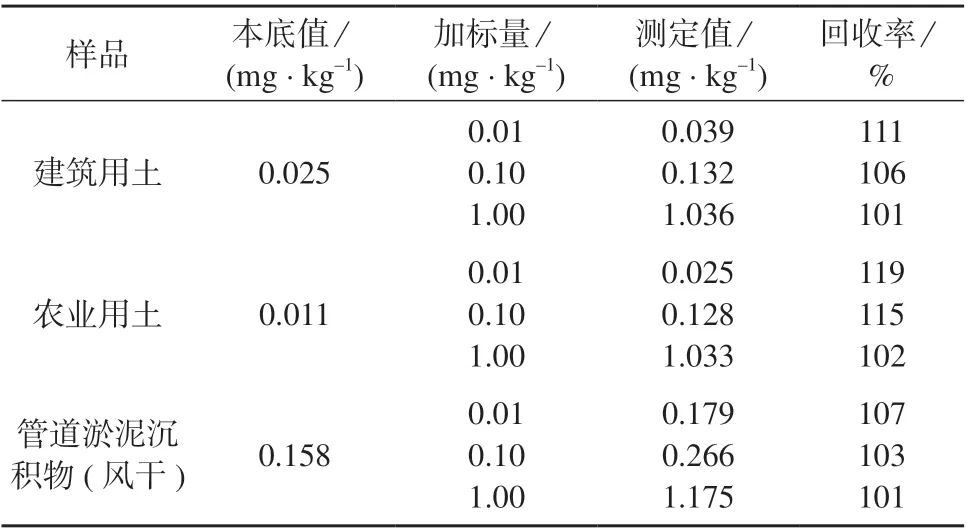
表7 加标回收试验结果
2.7 方法比对
本方法与部分文献方法比对结果见表8,由表8可知,本方法的准确度和精密度均满足要求,并优于文献报道的方法。经优化后的水浴法消解时间与微波法相同,且不需赶酸,适合于高通量样品的处理。
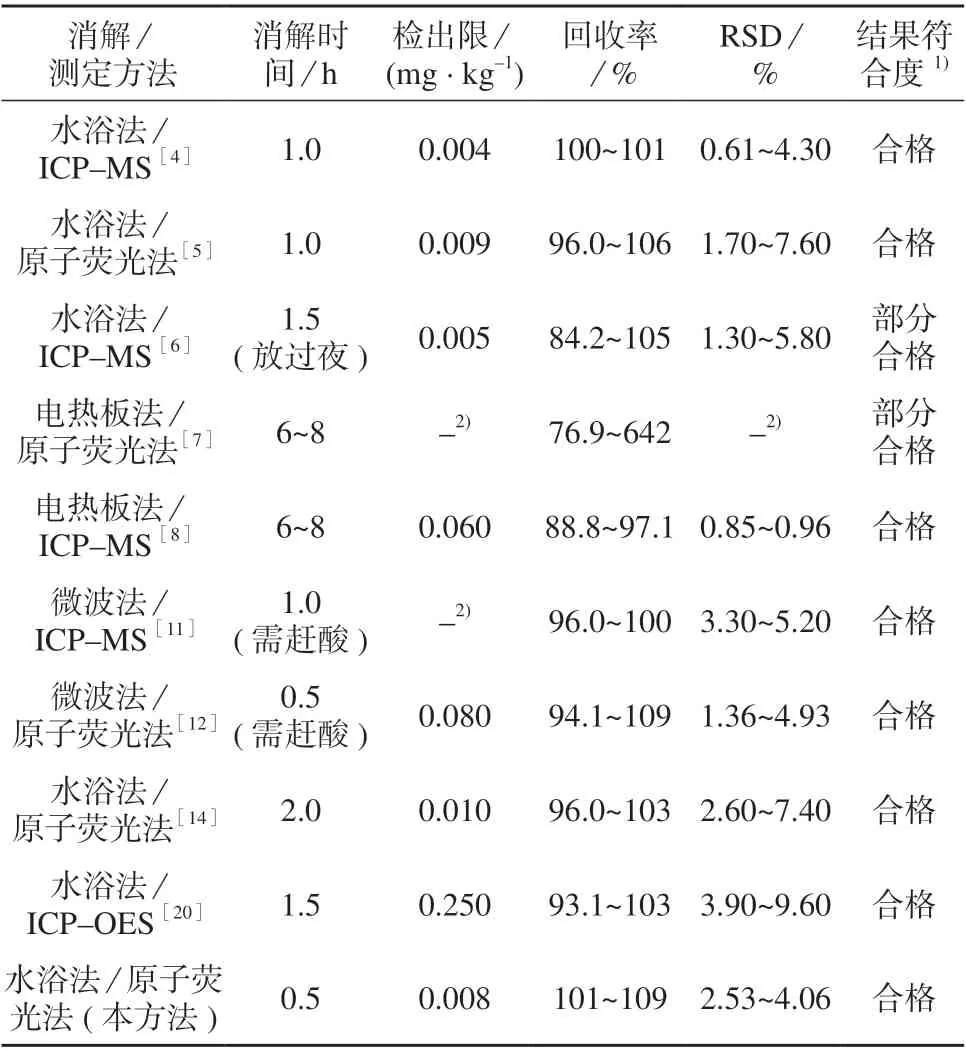
表8 本方法与文献方法比对结果
3 结语
以国家一级土壤成分分析标准物质GSS–16,GSS–17,GSS–23 与台州农业、建筑用土和管道淤泥沉积物样品为试验对象,采用水浴法前处理样品,用原子荧光法测定土壤和沉积物中痕量铋。探讨了13 种混合酸消解体系对样品痕量铋测定结果的影响,筛选得到检测痕量铋含量的最佳实验条件。该方法可用于高通量环境污染检测行业,具有良好的准确性和精密度,且水浴法不需赶酸,消解时间短,同一消解管即可完成消解、定容和上机分析全部操作,避免了多步骤转移带来的测定误差,提高了分析结果的准确性。
