制药装备中计算机系统的质量控制分析
2020-11-19张雄师
张雄师
摘要:计算机技术在制药领域的应用表现为:使用计算机系统控制制药装备,实施药物自动化生产。基于此,文章结合GMP指南在计算机系统中的应用方式,以无菌配液自动化系统为例,论述制药装备中计算机系统的质量控制方法与要点,为制造厂合理规范应用计算机系统提供实践帮助。
关键词:制药装备;计算机系统;GMP指南
前言:
在制药厂的生产中,制药装备表现出自动化、系统化发展趋势,如配液系统、净化系统等,均由两个及以上单体设备组成,为智能化、现代化制药提供技术支持。国家药品生产质量规范中明确指出,在药品生产的各个环节,均需实施GMP验证与确认,计算机系统也处于药品生产流程内,需对其进行验证处理。
一、GMP在计算机系统中的应用方式
GMP是指药品生产质量管理规范,用于约束制药厂的药品生产工序,保障药品安全与质量。在药品生产技术不断创新升级背景下,GMP不断修订与完善,制定了GAMP指南,对制药装备使用的计算机化系统进行验证与管控。目前GAMP指南已颁布第五个版本(GAMP5),该版本从质量风险管理角度入手,对计算机化系统在制药装备中的应用进行全生命周期的分析。目前制药厂使用的制药装备计算机系统有定制软件与可配置软件两类,前者的使用更为广泛。
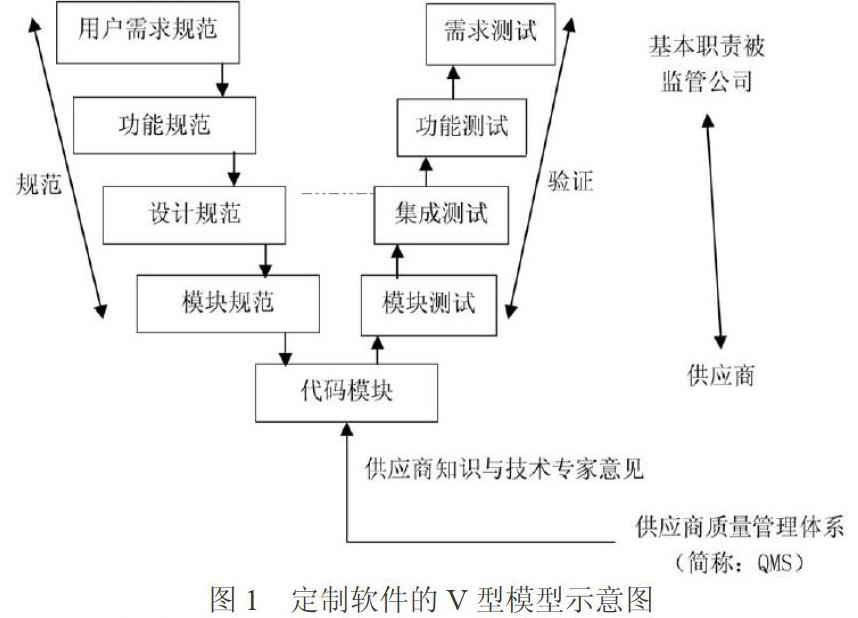
在GAMP指南中,计算机化系统的验证遵循V型模型,定制软件的V型模型如图1所示。可见,V型模型包括两部分,分别为规范与验证,规范的内容相对固定,包括用户需求规范、功能规范、设计规范及模块规范;验证的内容并不固定,由计算机系统决定,常见验证内容包括需求测试、功能测试、集成测试及模块测试[1]。
二、制药装备中计算机系统的质量控制要点
通过上述分析可知,制药厂需遵循GAMP5的要求,对制药装备中使用的计算机系统进行管理,规避质量风险,保障药品安全与质量。为明确制药装备中计算机系统质量控制要点,本文以常见的制药装备——无菌配液系统为例,结合其特点及GAMP5的要求,按照规范流程开展质量控制工作,为制药厂提供成功经验参考。
(一)整体验证架构
案例无菌配液系统选择西门子300系列PLC作为控制设备,与现场HMI触摸屏及远程I/O站配合使用,共同构成无菌配液系统的控制模块;在仪表设备模块配置称重系统;在监控模块配置PC上位机及打印机。在无菌配液系统的V型模型中,验证流程为计划→规范→配置和编码→确认→报告。
(二)计划验证流程
在计划验证环节,技术人员根据GAMP5中关于可增减生命周期和质量风险管理的条文,对无菌配液系统的软件与硬件设施进行分类处理,根据类别选择相应的验证模型与工序,选择最佳验证方式。在软件分类中,PLC属于5类软件,符合GAMP5中定制软件要求,可将其归属于定制软件范畴,按照上文中V型模型进行验证[2]。
(三)规范验证流程
根据定制软件的V型模型,规范验证内容包括用户需求规范、功能规范、设计规范。
在用户需求规范验证中,需掌握制药厂对无菌配液系统的需求,以此为基础,开展系统设计及相关验证活动,使供应商与质量风险评估更为准确,深化对无菌配液系统工艺流程的认识,保障药品生产质量。
在功能规范验证中,技术人员需了解无菌配液系统的功能要求,以此制定相应的功能规范,要求功能规范的内容准确体现系统功能要求,确保无菌配液系统符合制药要求。在该项工作中,技术人员可通过顺控伪代码或程序流程图,使无菌配液系统的各个模块与制药工序相对应,提高功能规范的合理性。
在设计规范验证中,验证内容包括以下三部分:(1)硬件设计,技术人员需结合无菌配液系统的运行网络、控制系统及操作界面,准确定义无菌配液系统各个硬件设备的设计规范;(2)软件设计,技术人员可将功能规范为基础,对PC软件、PLC程序、数据处理软件及HMI软件进行定义,定义内容包括软件框架与结构;(3)控制功能软件设计,在无菌配液系统中,不同软件模块承载不同功能,需结合功能规范,进行定义与编程,涉及模块的接入、组态及编码环境等内容,保障功能模块设计的合理性及准确性。
(四)验证处理与管理
根据GAMP5规定的内容,在规范验证完成后,需对验证内容进行如下处理,并开展风险管理工作,实现计算机系统的全面质量控制。
第一,配置与编码。在配置与编码环节,主要完成以下工作:(1)全面整合软件与硬件生成的相关信息,如开发工具、代码标准及命名等;(2)做好组态管理与版本控制工作,技术人员应用版本控制工具,对软件编程进行控制,确保其符合标准规范;(3)对于软件开发中应用的文本语言,进行代码审核。为避免代码审核不合格,技术人员可选择图形化编程手段,替代文本语言。在通过代码审核后,即可进行软件的开发测试,该工作也需遵循相关规范。
第二,验证确认。在无菌配液系统验证确认工序中,主要进行安装确认、运行确认、性能确认三项工作。其中,性能确认为非必须部分,前两者为必须确认的内容。在安装确认中,技术人员需根据相关规范,选择合适检测方法、技术,验证无菌配液系统的软件与硬件是否正确、合理安装;在运行确认中,技术人员需结合功能规范,对安装完成后的无菌配液系统进行调试及试运行,观察试运行期间无菌配液系统的各项参数与功能是否符合制药厂生产需求;性能测试是对无菌配液系统的各项性能进行检测,如加热性能、冷却性能、清洗性能,确认方法为检测实验,技术人员需按照规范实验进行确认。以加热性能为例,其验证实验如下:向罐内添加80L的注射用水,开启蒸汽阀门,记录水沸腾所用的时间和最终温度。
第三,验证报告。在无菌配液系统试运行确认无误后,即可进行验证报告,结合无菌配液系统验证全过程及验证结果,按照规范格式填写验证报告,并将清单移交,完成验证。
第四,风险管理。在提交验证报告后,需结合验证内容,遵循ICH:Q9和ISPE等规范与理念,开展风险管理工作,将风险管理渗透于无菌配液系统验证全过程,及时发现无菌配液系统存在的缺陷与不足,防控质量风险。
结论:
综上所述,在制药装备计算机系统质量控制工作中,工作人员需遵循GAMP5的要求。通过本文的分析,技术人员需结合计算机系统的特点,对其进行分类按照GAMP5规定的模型,对计算机系统进行验证分析,如计划验证与规范验证,并做好配置与编码、确认、报告与风险管理等工作,保障制药装备规范有序运行,提高药品质量。
參考文献:
[1]秦昆明,李伟东,张金连,等.中药制药装备产业现状与发展战略研究[J].世界科学技术-中医药现代化,2019,21(12):2671-2677.
[2]石正国.践行GMP质量风控 期待制药装备“鸟枪换炮”[N].医药经济报,2019-02-18(007).
