样本采集与处理条件对中华按蚊转录组测序结果的影响*
2020-11-19耿合员孙立新聂国嫒胡双双杨庆贵
耿合员 何 江 王 渊 孙立新 聂国嫒 胡双双 杨庆贵**
(1.江苏省检验检疫科学技术研究院, 江苏南京 210019; 2.江苏国际旅行卫生保健中心, 江苏南京 210019; 3.陕西国际旅行卫生保健中心, 陕西西安 710068)
中华按蚊Anophelessinensis是严重威胁人类健康的病媒生物,野外种群数量大,滋生环境复杂,适应能力强,能够通过叮咬宿主传播包括疟疾、马来丝虫病、流行性乙型脑炎等多种传染病,因此长期以来是预防医学的重点监测防控对象(Zhangetal., 2015; Fengetal., 2017; Fangetal., 2018)。随着测序技术应用的普及,以转录组测序为代表的基因研究逐渐成为中华按蚊等害虫的主要研究手段之一。目前常规意义上的转录组测序是指基于Illumina等短读长平台对样本mRNA的序列获取,结果分析包含不同基因mRNA的数量、类型与表达量等信息。基于转录组数据库的比较能够解析目标物种包括不同发育阶段、生理反应、胁迫应答等条件差异下参与的基因及其可能的作用机制。对于中华按蚊基于不同时空与发育、胁迫条件下的比较转录组分析已在此物种吸血分子基础、消化代谢与免疫以及抗药性机理等方面取得了大量进展,为深入理解其生理生态基因表达调控机制提供证据支持(Sunetal., 2017; Liuetal., 2018; Yanetal., 2018; Zhouetal., 2018)。但是,转录组测序技术的成熟也伴随着新问题的出现,其中样本不同采集与保存方法所导致的差异凸显,成为了影响测序与分析结果的重要因素(Shenetal., 2018; Sonetal., 2018)。以中华按蚊为例,其个体藏身隐蔽且十分脆弱,野外活体采集具备难度,部分样本采集完拿到实验室时已死。而且,由于野外采集的实验条件通常有限,因此样本在送达实验室安全保存前通常也会发生不同程度的RNA降解。此外,中华按蚊体内常具有已吸取的宿主血液,从而对样本的RNA提取造成干扰。这些干扰因素均会影响样本构建测序文库中mRNA的丰度,降低后续分析的准确性。
鉴于此,本研究拟对中华按蚊在吸血、存活状态及投入RNA贮存液前的冷冻时间等变量下转录组测序结果差异进行比较分析,分析这些变量下组装的完整性,鉴定检出基因的组成及表达量差异,以及这部分差异基因主要的功能。本研究结果将从方法学角度,为之后中华按蚊的转录组测序研究在样本采集与保存方面的策略优化,以及有效评估现有转录组数据有效性等方面,提供详尽数据支持。
1 材料与方法
1.1 样本采集
中华按蚊样本采集自云南瑞丽一养牛场。依据本研究的目的,共采集4类样本,样本均为雌性,分别为未吸血活体、吸血活体、吸血活体在-20 ℃冷冻20 min、吸血死样本在-20 ℃冷冻20 h。详细的样本分组如表1所示。

表1 中华按蚊转录组测序样本信息
1.2 RNA样本提取与转录组测序
使用天根试剂盒提取各组样本的总RNA,操作方法参考试剂盒说明书。每组选择5个个体,混样提取,提取出的RNA样本使用Bioanalyzer平台进行检测合格后,用于转录组测序。转录组文库构建与测序委托深圳华大基因公司完成,采用BGISEQ-500平台,每组3个生物学重复,4组共计12个样本,每个样本测序至少6 Gb。
1.3 数据处理与分析
测序原始数据使用Trimmomatic v0.33软件(Bolgeretal., 2014)过滤包含接头、未知碱基N含量大于5%等低质量序列。所有过滤后的清洁标签(Clean reads)用于之后输入Trinity v2.11.0软件(Grabherretal., 2011)合并组装,然后使用Tgicl v2.1软件(Perteaetal., 2003)对转录本进行聚类去冗余得到最终转录组基因数据集Unigene。Unigene的质量使用BUSCO v3(Benchmarking Universal Single-Copy Orthologs)软件(Seppeyetal., 2019)中单拷贝直系同源基因(Single-Copy Orthologs, SCO)完整性比例进行评估,评估采用昆虫(insecta)共有SCO基因数据库。使用TransDecoder v5.3.0软件(Grabherretal., 2011)识别Unigene中的编码区域(Coding domain sequence, CDS),识别结合Blast 2.2.28软件(Camachoetal., 2009)比对SwissProt数据库(McMillanetal., 2008)和Hmmerscan软件(Chaietal., 2019)搜索Pfam 数据库中(Finnetal., 2014)蛋白同源性的结果,以获得更完整编码区域。
功能注释通过将组装得到的Unigene在常用功能数据库中依据序列相似性比对获得KEGG(Kanehisaetal., 2017)、GO(The gene ontology resource)、NR、NT(Bensonetal., 2018)、SwissProt(McMillanetal., 2008)、Pfam(Finnetal., 2014)和KOG(Mudado Mdeetal., 2006),此过程中采用的软件分别为KAAS(Moriyaetal., 2007)(KEGG)、Blast2GO basic(Conesaetal., 2005)(GO)、Blast v2.2.28(Camachoetal., 2009)(NR、NT、SwissProt、KOG)、Hmmscan v3.2.1(Chaietal., 2019)(Pfam)。
使用 RSEM v1软件(Lietal., 2011)将各个样品与Unigene进行比对,计算基因表达水平。R中deseq软件包(Andersetal., 2010)被用于计算不同样本组(重复样品归为一组)的差异表达量分析,使用每百万碱基检出片段数量(Fragments per Kilobase per million,FPKM)值作为衡量表达量差异的参数。差异倍数达到两倍以上并且二轮校准后的P-value(Q-value)≤ 0.001 被用来筛选显著差异表达基因。差异基因的GO、KEGG富集分析使用R软件中的phyper软件包(Berlivetetal., 2007),Q-value≤ 0.05的功能视为显著富集。结果的可视化通过R软件中的GG-plot2软件包(Maag, 2018)完成。
2 结果
2.1 转录组组装结果
转录组组装结果所得的Unigene基因数据集及其相关功能注释信息是之后比较分析的基础。本研究所选择的12个中华按蚊样本共测得75.58 Gb 数据,平均每个样本测序6.30 Gb。最终去冗余从头组装结果包含48 775个Unigene。序列总大小为 68.1 Mb,平均长度为1 395 bp、N50值为2 349 bp,整体GC含量为47.75 %。进一步从这些Unigene中检测出28 265个编码序列CDS,同时预测出5 263个编码转录因子。综合统计Unigene比对到不同基因功能数据库的结果,显示共有35 567(72.92%)个基因能够获得功能注释信息。其中各数据库分别注释了33 330(NR:68.33%)、19 966(NT:40.93%)、25 033(SwissProt:51.32%)、24 512(KOG:50.26%)、26 293(KEGG:53.91%)、26 704(GO:54.75%)以及25 756(Pfam:52.81%)个基因。
基于统一的Unigene数据集,研究获取了各样本中所包含的基因及其表达量。表达量定量结果显示,各组间全部基因表达量分布范围接近,log10(FPKM+1)的值主体介于0到1之间,离散点较少(图1)。基因表达密度统计显示各样本表达分布高度重叠,只存在一个单一的峰(图2)。这些结果说明构建的Unigene数据集在解析各样本时的结果可靠,可用于后续比较分析。
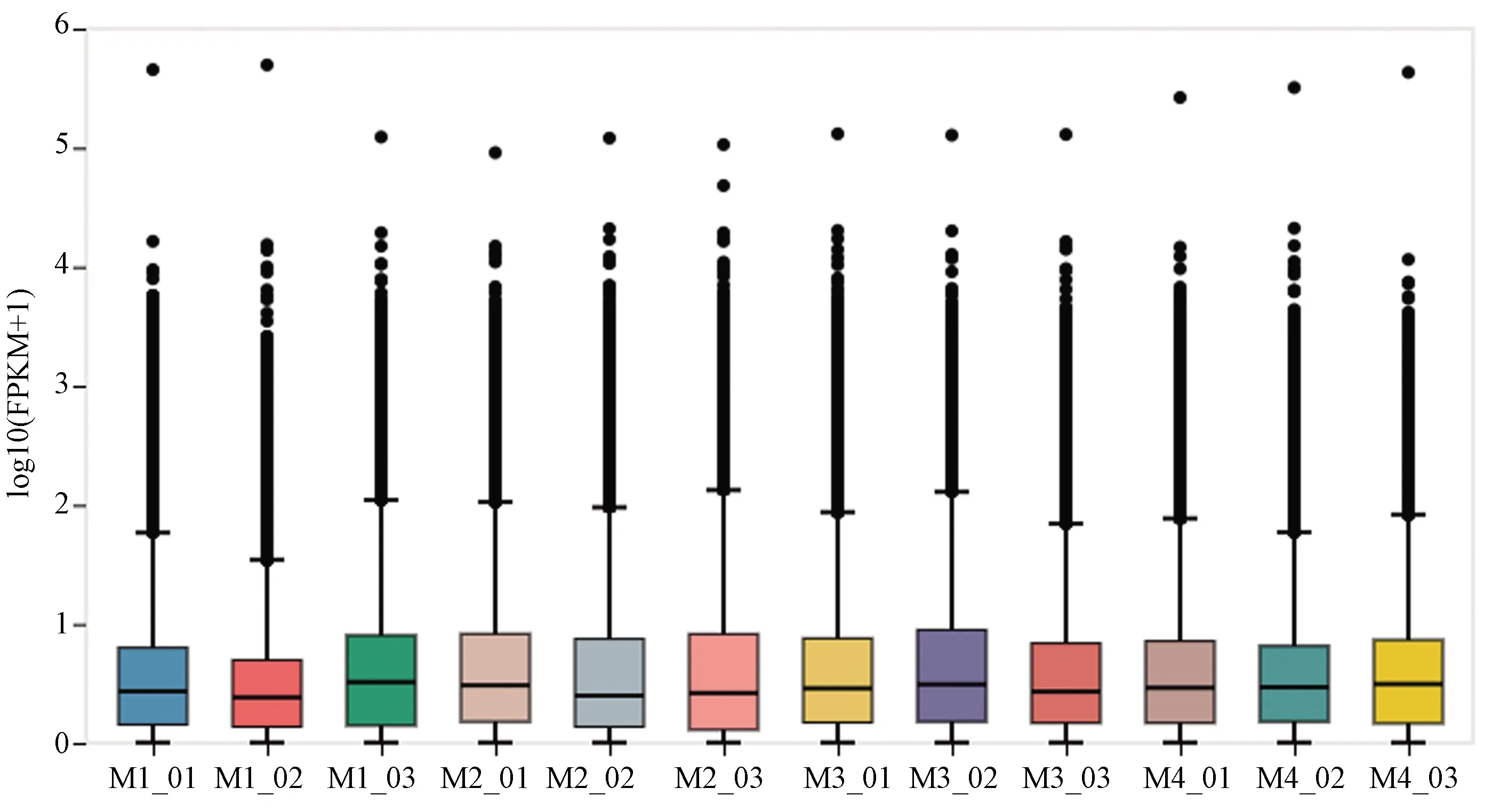
图1 表达量箱线图
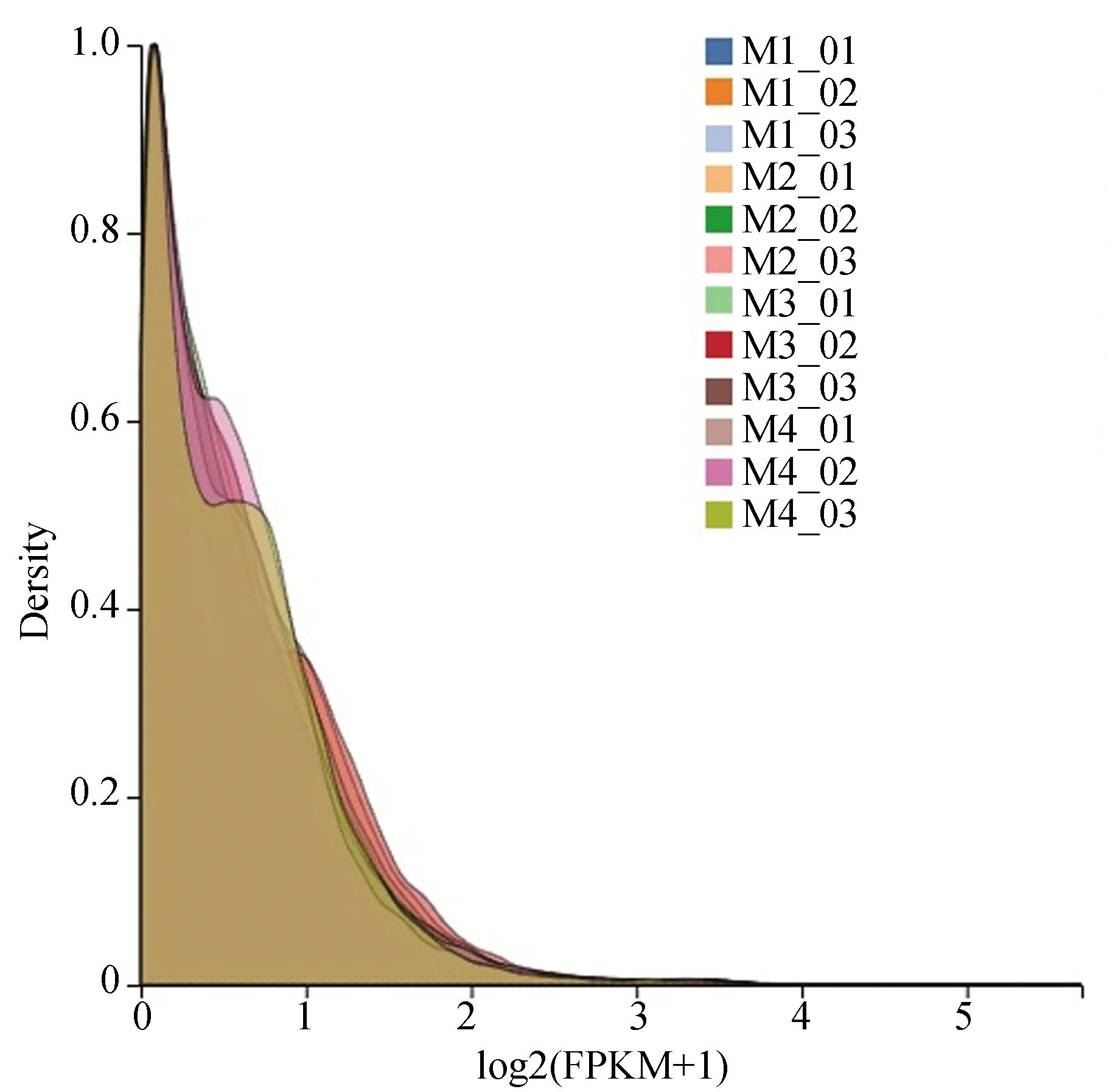
图2 表达量密度图
2.2 中华按蚊转录组组间组装质量比较
研究首先比较了各组样本相对于整体的组装完整性。结果显示,整体Unigene的SCO完整性极高,达到了98.7%。各组间比较结果显示全部样本的完整性均低于整体,即各组均有部分基因未能检出。之后,组间比较也发现明显差异,其中M1组与M4组的SCO完整性低于M2组与M3组,而M1组2号重复样本与M4组3号重复样本的完整SCO基因比例甚至低于60%,存在大量片段及未能检出的缺失的SCO基因(图3)。该结果说明至少从SCO抽样水平,各组检出的基因数量及质量有区别。
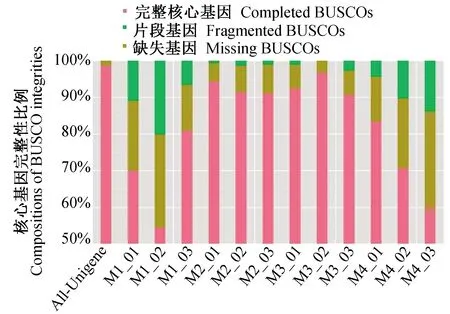
图3 组装结果SCO基因完整性示意图
2.3 组间差异基因鉴定
之后进一步从全转录组完整Unigene水平鉴定并比较了各组检出基因的差异。结果显示,四组共有的Unigene为 28 183条,仅占全部的57.78%(图4)。从韦恩图上可知,M3与M4组中检出基因较少是导致共有基因占比较低的原因,二者分别只检出33 267与33 058个基因。而M1与M2组检出基因数量更多(分别为45 887 与44 478个),二者共有42 584个基因,占全部的87.31%(图4)。进一步,为了揭示M1与M2组所多检出的基因的功能分布,本研究对在M3与M4组中均未能检出而M1与M2组均检出的11 358条基因的功能进行了富集。结果显示,GO与KEGG富集的通路均与基础代谢机制相关(图5~6)。如GO中最显著富集基因为发生在核内(Nucleus)的核酸结合(Nucleic acid binding)与锌离子结合(Zinc ion binding),为基础代谢中最常应用的分子功能(图5)。而KEGG中最显著富集的基因主要参与了核糖体生物合成(Ribosome biogenesis in eukaryotes)、谷胱甘肽生物合成(Glycosaminoglycan biosynthesis)、基础转录因子(Basal transcription factors)等基本物质代谢途径(图6)。
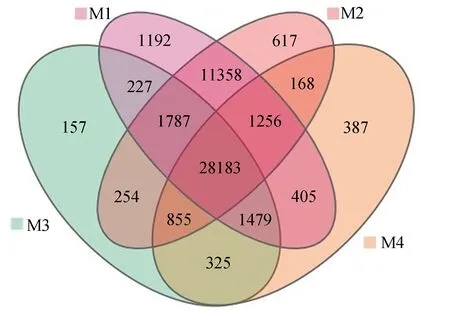
图4 转录组组间基因韦恩图
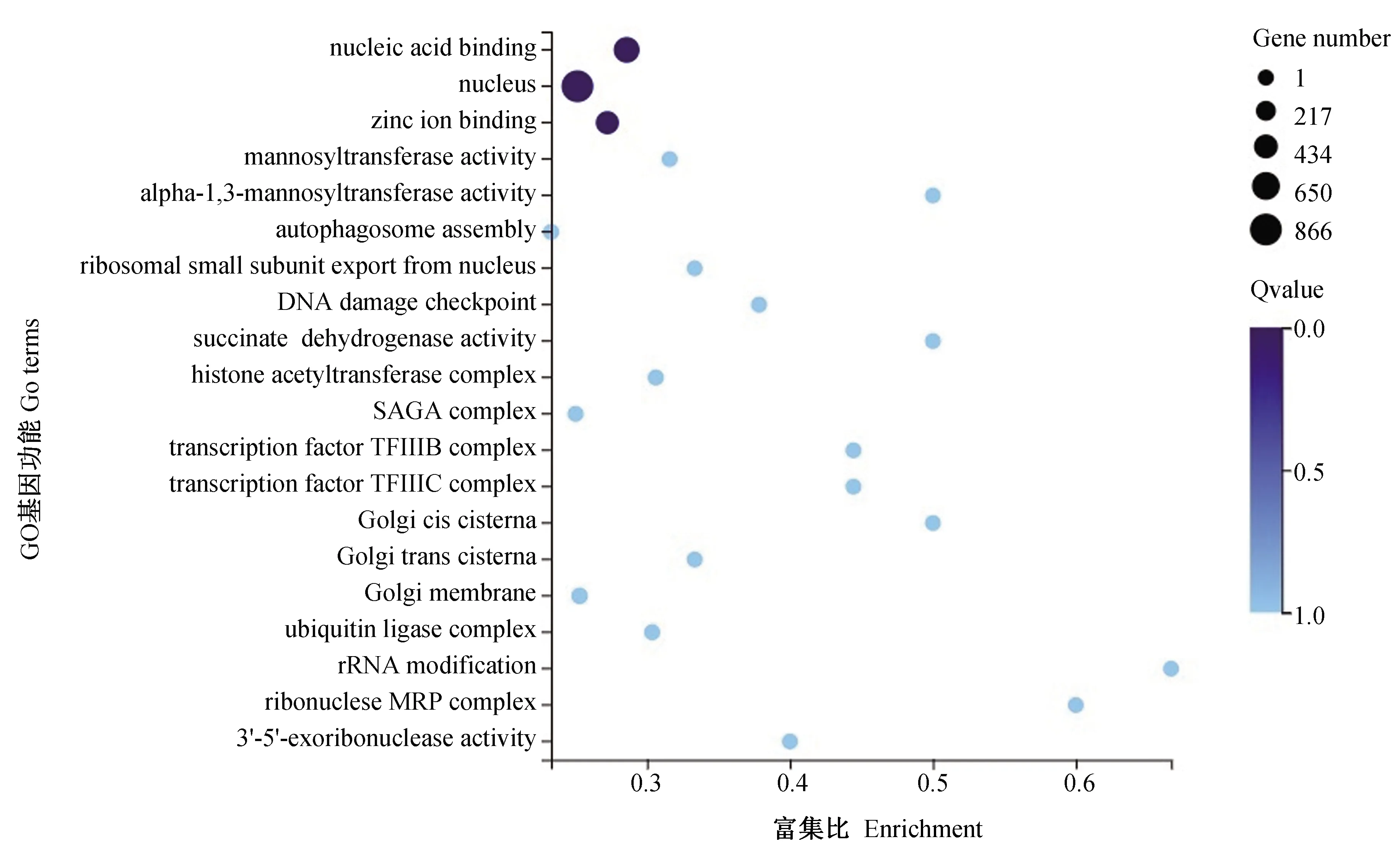
图5 GO类群功能富集图
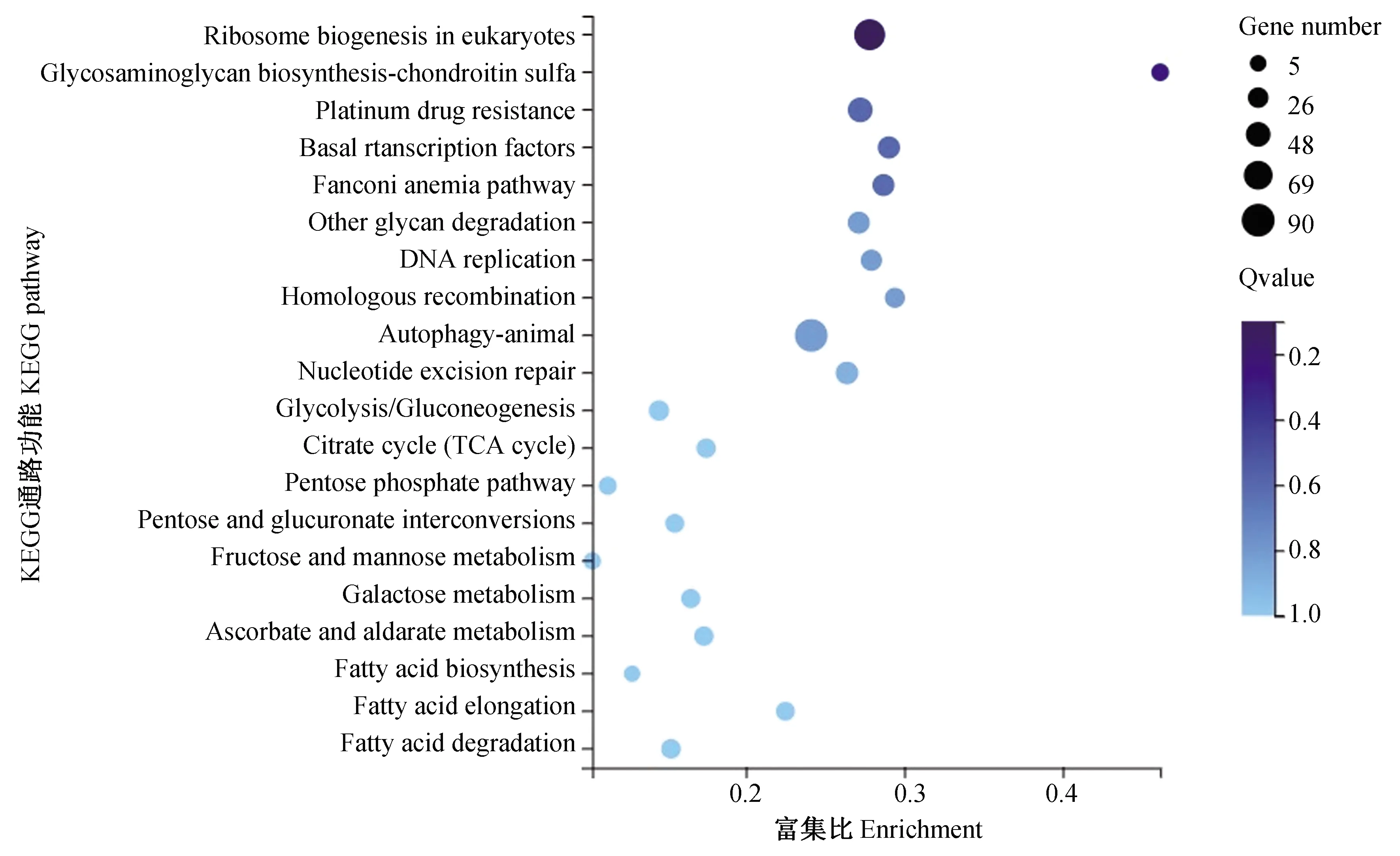
图6 KEGG通路富集图
2.4 组间共有基因差异表达水平
对于所有组均检出的基因,研究比较了其表达量以分析不同变量在解析表达水平层面上的影响。结果显示,任一两组间均存在大量差异基因,发生显著上调与下调的基因总数均超过10 000 条,超过全部共有基因(28 183条)的30%。具体而言,研究发现M3与M4组间的差异表达基因数量最少,且上下调接近;M1与M2组相比上调基因多于下调,而其余组比较均为下调基因多于上调基因;M1与M3组发生显著调控的基因数量最多,且上调与下调基因数量均为各比较组中最高(图7)。
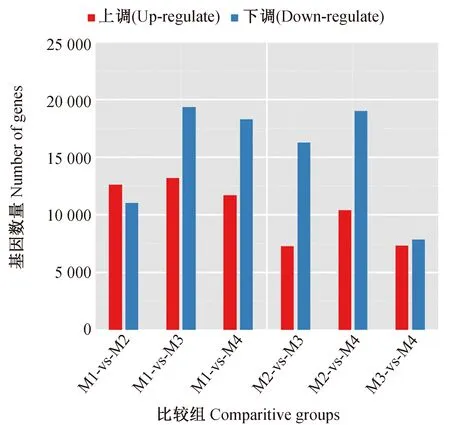
图7 差异表达基因统计图
通过对各组差异表达基因分别进行GO功能富集以比较其组成与功能上的差异,发现这些基因几乎不存在组间功能重叠,表现出强组间特异性特征(图8)。对于M2组与M3组而言,其差异表达基因的作用位置甚至都与其他组不同,该组基因主要作用于胞外区域(extracellular region),而其他组差异表达基因则主要作用于核内。该组中与能量代谢相关的通路(ATP synthesis)也同样被富集出来。M1组与M2组中与细胞代谢活性相关的通路发生显著富集,如DNA复制(DNA replication)与RNA结合(RNA binding)等。M3与M4组比较,没有获得有实际生物学意义的GO分类。M1与M4组相比,富集了多个发挥免疫与解毒相关的分子功能,包括内肽酶活性(Endopeptidase activity)与去氧化活性(Oxidoreductase activity)等。这些功能均反映着各比较组中所对应的变量对基因表达量的影响,提示不同的保存与处理方法对于样本RNA质量的干扰明显。

图8 差异表达基因GO类群富集图
3 讨论
样本的RNA是转录组测序的物质基础,其质量决定了结果的可靠性。在本研究中,我们以中华按蚊这一需监控的且具有复杂样本采集与保存过程的病媒生物为材料在4种不同条件变量(非吸血活体、吸血活体、吸血活体短期冷冻、吸血死体长时间冷冻)下的转录组测序结果差异进行了分析与比较。本研究依次从转录组组装水平、检出核心直系同源基因比例、整体检出基因数量差异及功能富集、共有基因的表达差异及功能富集等4个层面进行了详尽的量化评估。
经结果整合,研究发现吸血活体为最优样本,其3个重复样本的核心基因完整性均超过90%,检出基因总数占完整Unigene数据库的比例也达到了44 478个,占全部的91.19%。这与研究预期的非吸血活体样本为最优存在一定偏差,即吸血样本中存有的宿主血液中的核酸对转录组测序样本的干扰影响十分微弱。进一步推测其背后的原因,研究首先排除了多检出基因为宿主引入的可能性,即吸血活体与非吸血样本相比较其检出的基因具有高度重叠,之后基于两组间差异表达基因的功能富集结果主要为能量代谢相关,推测吸血样本具有更活跃的代谢水平可能导致其基因表达丰度更高,从而更容易被转录组测序平台捕获。之前认为的宿主外源污染,其可能在样本捕获时便已降解,或在之后组装前的数据过滤阶段被清除,对测序结果不构成实际影响。
与吸血组相比,非吸血活体样本检出基因数量最多,达到45 887个,占总数的94.01%。但是,该样本的核心直系同源基因完整性却极低,平均仅为63.3%。从核心基因的构成看,推测该样本可能存在组装问题,即测序对基因的覆盖不完整,呈现出碎片化特征。导致该情况出现的原因从样本生理状态的角度较难找到合理解释,因为如果是个体虚弱或降解的原因,其差异表达基因与检出基因数量应该均明显少于目前鉴定的状况。于是,研究只能暂且推测该样本是在建库过程中mRNA捕获时便出现了缺失,具体根源还有待于之后通过其他测序技术方法进行探究。
对于样本的捕获状态,研究发现采集时立即处死对于转录组测序的样本影响较小。与活体样本相比,死亡个体样本检出基因最少,且具有大量差异表达基因,即存在明显降解。然而,出乎意料的是,就核心基因完整性而言,死亡样本并未发生断崖式下降,其与非吸血活体样本接近,存在大量片段化基因。除此之外,在检出的基因中,其与活体短期冷冻20 min样本间具有广泛重叠,而与其他活体组鉴定出的差异表达基因中,活体样本中发生下调的基因数量均多于上调,即死体检出基因表达表现出受刺激应答反应的特征。这些结果与通常认为的死亡个体后mRNA会快速完全降解的认知不符,推测是由于本研究所选择的个体是采集时才处死的,采集之前此个体的躲避行为可能导致了其表现出刺激应答特征。该推测也一定程度上得到了差异基因功能富集的支持,本研究中的死体组与其他吸血组相比光传导(Phototransduction)均被显著富集,与非吸血组相比,则有多个解毒与消化相关的基因发生表达改变,均符合各组样本间的生理差异。
此外,研究还发现冷冻处理对于转录组测序样本的影响极为显著,其严重性远超本研究中所讨论的其他变量。在研究所设计的两组经过冷冻处理的样本中均出现了极为明显的降解,表现为检出的基因数量较未经冷冻组减少了均超过10 000 条,占全部Unigene的总数超过20%。而通过对这部分未检出的基因(11 358条)进行功能富集也证实了广泛降解的发生,即这部分基因功能参与了包括核酸结合及生物合成等多种基础物质代谢机制。之后,基于冷冻20 min与冷冻20 h的样本基因检出总数相近这一结果,研究认为冷冻处理导致的样本降解主要发生在处理前期,且很可能在20 min内,而随着冷冻时间的增加,降解过程逐渐变得缓慢。
综上所述,本研究通过对中华按蚊非吸血活体、吸血活体、吸血活体短期冷冻、吸血死体长时间冷冻下的转录组测序结果差异进行分析与比较,发现吸血活体为最优样本。各变量间吸血可能引入的污染对样本的影响最小,样本捕获导致的死亡与冷冻均会导致样本降解,其中冷冻时间对样本质量的影响极大,可在短时间内造成样本严重降解。
