烟草青枯病抗性的全基因组关联分析
2020-11-04何斌彬耿锐梅杨爱国
何斌彬,耿锐梅,杨爱国,任 民*
(1.中国农业科学院烟草研究所,青岛 266101;2.中国农业科学院研究生院,北京 100081)
烟草青枯病是由青枯雷尔氏菌(Ralstonia solanacearum)侵染引起的一种土传细菌病害,可侵染50 多科的数百种植物[1],在世界范围内均有发生,近年在我国危害程度逐渐上升,且发病区域有向北蔓延的趋势[2],已成为我国烟草生产中的主要病害之一。
音乐教学需用到很多教学器材,往往因为学校对其课程的不重视,导致投入资源较少,产生音乐教学器材不完善的现象。这种现象限制着教师的教学水平及学生的体验水平。针对这一问题,学校应全面认识素质教育的重要性,对待音乐教学环节适量加大资源投入,确保学生的教学体验,帮助学生进行全面的素质教育建设。
已有研究表明烟草青枯病抗性遗传较为复杂[3]。利用晾晒烟[4]、香料烟[5]及烤烟[2]进行的青枯病抗性遗传分析表明,在不同烟草类型以及同类型不同品种间的青枯病抗性遗传规律差异均较大,来源于同一抗源亲本的品种之间,其遗传规律也并非完全一致,但基本认为烟草青枯病抗性为多基因控制且具有加性效应。有关烟草青枯病抗性QTL 的定位研究开展较早[6],利用AFLP、SSR 等分子标记,在不同的分离群体内,鉴定到了多个青枯病抗病QTL 位点;利用MELP 和SSR 标记,探索了基于关联分析的青枯病抗性位点发掘,并取得一定研究进展[7-8]。但由于上述研究普遍采用的是基于酶切和PCR 的二代分子标记技术,其标记数量有限,定位区间较大,且缺乏基因组信息支撑,限制了研究结果在抗病基因克隆和抗病分子标记辅助选择育种中的应用。
近年来,随着高通量测序技术的快速发展,单核苷酸多态性(Single Nucleotide Polymorphism,SNP)和插入缺失突变(Insertion-Deletion,InDel)等新一代分子标记在动植物遗传研究中得到了广泛的应用。与SSR 等传统分子标记技术相比,SNP标记具有数量多、密度高、分布广泛等优势[9]。基于简化基因组测序的RAD 技术(Restriction siteassociated DNA sequencing,RAD-seq),在实现基因组全覆盖的同时,还能大幅降低测序成本[10],非常适合烟草这类基因组庞大的作物。本研究以219 份国内外表型变异丰富、遗传多样性较高的烤烟品种组成的自然群体作为研究材料,利用RAD 简化基因组重测序技术,对烟草青枯病抗性进行全基因组关联分析,发掘与抗性变异显著关联的区段,并预测抗病候选基因,为开展烟草青枯病抗性分子育种奠定基础。
随着航天发射场规划建设水平的不断提高,发射场的发展进入了一个新时期,必然呼唤与之适应的设备体系.发射场机械设备推进“三化”(通用化、系列化、组合化),实现特种设备的装备化管理成为了发射场建设的新目标.模块化设计的总体目标是以较少的资源满足多样化的需求,将可靠性、维修性和保障性为核心的先进理念应用于设计阶段,实现发射场机械设备的顶层规划和一体化规划.依据变型设计的有关原理,进行通用化设计,运用功能模块置换,使特种机械设备种类大大减少,提高设备的互换性与适应性,促进设备的装备化保障管理,从而推进实现装备的通用化、模块化、系列化.
1 材料与方法
1.1 供试材料
在关联分析的基础上,利用烟草基因组数据库,根据上述对烟草烤烟品种群体的LD 衰减距离分析结果,在与关联峰值SNP 位点上下游各256 kb 的LD 区间内共筛选到18 个基因,详细信息见表2。根据基因功能注释信息及相关研究报道,推测Ntab0717150、Ntab0717130、Ntab0080360和Ntab0080340这4 个基因可能与青枯病抗性相关。
1.2 简化基因组重测序及SNP 鉴定
通过RAD-Seq共发掘到高质量SNP位点384 904个,筛选到非缺失个体不少于190 的SNP 位点共338 315 个,用于计算LD 衰减距离。通过滑动窗口法共获得16 914 526 组LD 数据,其中r2达显著水平以上的有2 071 065 组,占比12.24%。绘制LD衰减曲线(图2),以r2低于0.5 为临界值(SNP 位点间最高值1.0,初始窗口r2平均值0.92),在此条件下,烟草烤烟品种群体的LD衰减距离为256 kb。
1.3 病情调查
在移栽后60 d 发病期进行青枯病病情调查,参照行业标准GB/T 23222—2008,根据烟株茎部条斑和叶片凋萎情况将发病等级分为5 级,并根据病级计算每个供试品种的病情指数,根据供试材料的病情指数将品种抗病性分为6 类。
1.4 数据分析
关联峰值位点C17_23 977 382 在群体上的分型情况见表1,在群体上有AA、GG、AG 三种基因型,其中AA 型有23 个品种(10.5%),GG 型有188个品种(85.8%),AG 型有6 个品种(2.7%),缺失2 个品种(0.9%)。方差分析结果表明,等位变异A能够极显著(p<0.0001)大幅降低病情指数,是优异等位变异(图6)。
1.4.2 全基因组关联分析 利用TASSEL V 5.0[14]软件的MLM(PCA+K)模型进行全基因组关联分析,具体步骤同文献[12]。
供试品种的病情指数在各个区间均有分布,而抗病性主要分布在中抗、中感和感病3 个等级(图1),供试群体病情指数的偏度系数Skewness 为0.051,峰度系数Kurtosis 为-1.078,基本符合正态分布。共鉴定出8 个高抗青枯病的烟草品种,分别为Coker176、Special400、岩烟97、白色种、大青筋、Y-2、丸叶和K358,病情指数均为0。
1.5 候选基因预测
通过烟草基因组数据库(http://218.28.140.17/),获得关联区段内基因功能注释信息。
利用供试群体的青枯病抗性调查数据,与SNP群体分型数据进行了全基因组关联分析,由关联分析结果的Q-Q 图可知(图3),本研究所选用模型适宜,关联分析效果理想。以关联位点的p值小于1×10-6为多重检验(FDR)的显著性阈值,在此阈值之上共发掘到1 个关联SNP 区段(图4),其峰值SNP(C17_23 977 382)位于17 号染色体的23 977 382 bp 处(图5),p值为6.196×10-7,能解释14.29%的表型变异。
2 结果
2.1 供试群体的抗病性分析
首先,“营改增”政策对生产性服务业上市公司减税效应显著。由理论分析可知,“营改增”的减税效应主要取决于企业的外购商品及劳务能取得可抵扣进项的多少,实证分析表明,“营改增”后,企业外购商品及劳务增加,使企业总税负降低。同时,外购商品及劳务对企业税负下降的影响远大于“营改增”政策所带来的减税效应。
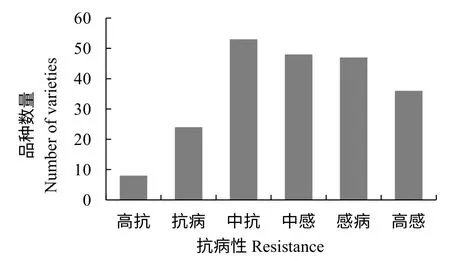
图1 供试群体抗病性分布Fig.1 The distribution of disease resistance in the test population
2.2 烤烟染色体LD 衰减分析
RAD 简化基因组重测序及SNP 鉴定与华大基因有限公司合作开展,参考基因组为烟草栽培品种红花大金元(V 4.0),建库酶切、高通量测序、SNP鉴定流程等参见文献[12]。
2.3 供试群体SNP 变异位点与青枯病抗性的全基因组关联分析
目前世界上大部分国家和地区都使用UTM投影。几内亚使用克拉克1880椭球,UTM投影,而我国已全面使用2000国家大地坐标系,国内通常使用高斯-克吕格(Gauss-Kruger)投影(以下简称高斯投影)。根据我国《工程测量规范(GB50026—2007)》:平面控制网的坐标系统,应在满足测区内投影长度变形值不大于2.5 cm/km的要求下(相对误差小于1/400 00),进行坐标系的选择。因此,必须熟悉和了解几内亚国家所使用的地球椭球及投影方式,才能建立好合适的坐标系统。
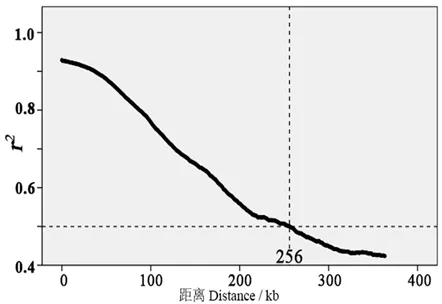
图2 烤烟品种群体的LD 衰减图Fig.2 LD attenuation map of flue-cured tobacco varieties

图3 青枯病抗性关联分析的Q-Q plot 图Fig.3 Quantile-quantile plot of association analysis of bacterial wilt resistance

图4 供试群体青枯病抗性的全基因组关联分析Fig.4 Genome-wide association analysis of bacterial wilt resistance in the test population
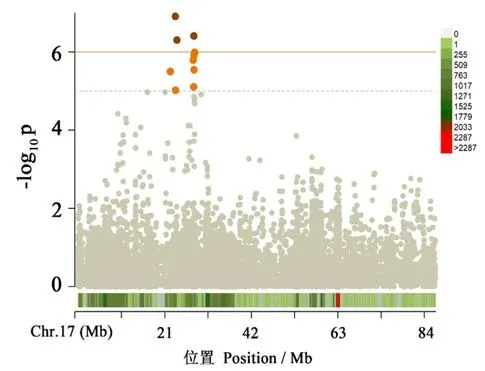
图5 17 号染色体上青枯病抗性关联分析的曼哈顿图Fig.5 Manhattan plot of association analysis of bacterial wilt resistance on chromosome 17
2.4 抗青枯病优异等位变异的筛选
1.4.1 连锁不平衡衰减距离计算 从全部SNP 位点中筛选非缺失个体不少于190 个的SNP 位点,利用Haploview 4.2[13]软件进行SNP 间的连锁不平衡(Linkage disequilibrium,LD)分析,计算50 个SNP以内两两位点间的LD 参数r2。用滑动窗口法统计r2的平均值,其窗口长度为36 kb,步长0.8 kb,绘制成LD 衰减曲线。
2.5 青枯病抗性相关基因
供试群体包括219 份国内外烤烟品种,由国家烟草种质资源中期库[11]提供。2016 年,供试材料种植于安徽宣城田间病圃,随机区组试验设计,2016年7 月调查田间自然发病情况。

表1 219 份供试材料分型结果Table 1 Typing results of 219 tested materials

表1(续)

图6 不同等位变异之间的表型差异Fig.6 Phenotypic differences between different alleles
3 讨 论
在之前的报道中[6],多数利用RAPD、AFLP 和SSR 等分子标记技术,通过连锁分析或关联分析的方法定位到一些与烟草青枯病抗性相关的位点,但存在标记非特异性、密度低和缺少基因组物理图谱信息等问题。得益于近年来快速发展的高通量测序技术,本研究与以往研究相比,实现了更高密度的基因组覆盖,同时关联到的SNP 位点具有明确的物理图谱位置,有助于研究的深入开展和互相验证。本研究通过全基因组关联分析策略,在烟草基因组内定位到1 个与烟草青枯病抗性变异显著关联的区段,峰值SNP 在17 号染色体的23 977 382 bp 处(C17_23 977 382),LAN 等[15]在烟草的17 号连锁群上检测到一个与烟草青枯病抗性相关的QTL(qBWR17a),能够解释30%的表型变异。本研究对烟草高密度SSR 遗传图谱利用电子PCR 及序列比对的方法,进行了SSR 标记的红花大金元物理图谱定位(数据未展示),其中上述17 号连锁群基本对应17 号染色体,因此本研究与LAN 等[15]研究结果较为一致。

表2 参考基因组第17 号染色体关联区段内18 个基因的注释信息Table 2 Annotation information of 18 genes in the association region of chromosome 17 of the reference genome
植物基因组LD 衰减距离决定关联分析时所需的标记密度,在一定程度上也决定了候选基因的定位精度[16]。本研究发现,烟草烤烟品种群体的LD衰减距离为256 kb,与其他作物相比,属于LD 衰减较慢的物种,显著低于玉米(1~5 kb)[17]、籼稻(10 kb)[18]、芝麻(88 kb)[19],与番茄(256.8 kb,CER 群体)[20]、马铃薯(450~550 kb)[21]等茄科作物处于一个数量级。原因可能是烤烟品种群体经过长期的驯化选择,导致群体遗传多样性下降,位点间的相关性(连锁程度)加强,导致LD 衰减速度相对较慢[22]。
通过候选基因预测,共找到4 个可能与青枯病抗性相关的基因,分别为Ntab0717150、Ntab0717130、Ntab0080360和Ntab0080340。其中Ntab0717150和Ntab0717130与拟南芥AT5G15900基因同源,该基因为TBL(Trichome birefringencelike)基因家族成员,已有研究表明该家族基因参与细胞次级壁纤维素的合成和沉积[23],推测基因Ntab0717150和Ntab0717130可能通过促进细胞壁的合成进而增强植物抵抗青枯病的能力;基因Ntab0080360和Ntab0080340与拟南芥AT1G22710基因同源,该基因编码蔗糖转运蛋白(Sucrose transporter,SUT)[24],已有研究表明,烟草中NtSUT1基因的反义表达会大大增加黑斑病的感病率[25],番茄中SlSUT2基因的反义表达会使植株的菌根侵染率显著提高[26],推测基因Ntab0080360和Ntab0080340可能通过影响根系发育进而参与青枯病的抗病进程。后续拟利用基因敲除和过表达对上述4 个基因进行功能验证。
根据金融衍生产品的发展规律,欧式看涨期权可以分为资产看涨期权多头和现金看涨期权空头两个种类。资产看涨期权指的是,如果在到期执行日时,执行价格高于该金融衍生产品价格,期权执行价值为零;反之,期权执行价格等于其金融衍生标的资产本身面值,故该期权的价值表示为:e-r(T-t)STN(d1)=SN(d2)。
4 结 论
本研究首次利用SNP 标记和全基因组关联分析策略相结合的方法,对219 份烟草自然群体种质材料进行青枯病抗性分析,在烟草基因组内定位到1 个与青枯病抗性变异显著关联的区段,该区段峰值SNP 位于17 号染色体的23 977 382 bp 处,并在该区段筛选到4 个烟草青枯病抗病候选基因。该研究结果为后续青枯病抗病基因克隆及分子育种奠定了基础。
