ASAH1基因突变致脊髓性肌萎缩症2例并文献复习
2020-11-02康庆云廖红梅杨赛陈波杨理明
康庆云,廖红梅,杨赛,陈波,杨理明
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是由于脊髓前角及延髓运动神经元变性,导致近端肢体和躯干进行性、对称性肌无力和肌萎缩的神经变性病。SMA可由多种基因突变引起,但一般特指由位于染色体5q11.2-q13.3的SMN基因突变引起的常染色体隐性遗传病,称为5qSMA,而非SMN基因突变引起的SMA 称为非5qSMA。1979 年,Jankovic 和Rivera[1]首次描述并报道3例成人期起病的缓慢进展的肌无力患者,病程中出现肌阵挛癫痫,称之为脊髓性肌萎缩症伴进行性肌阵挛性癫痫(spinal muscular atrophy with progressive myoclonic epilepsy,SMA-PME),之后文献报道类似病例10 余例,基因检测均显示SMN 基因正常。Zhou 等[2]在2012 年确 定ASAH1 为SMA-PME 的致病基因,之后国内外文献中共报道12例通过ASAH1基因确诊的SMA-PME 病例[2-10]。2016 年,Filosto 等[10]首次报道2 例成人ASAH1 基因突变患者存在SMA 症状但不伴进行性肌阵挛性癫痫,扩展了ASAH1 相关SMA 的表型谱。2018 年10 月湖南省儿童医院神经内科诊断2 例ASAH1 基因突变相关SMA 但不伴进行性肌阵挛性癫痫患儿,现总结其临床特点并文献复习,以提高临床医师对本病的认识。
1 资料与方法
1.1 本家系临床资料
1.1.1 先证者 男,9 岁4 月,因“肢体乏力3 年余”于2018年9月至湖南省儿童医院神经内科就诊。患儿为第2胎,第2产,母孕期及出生史无异常,早期生长发育里程碑基本正常:7月独坐,8月能爬,1岁1月能独立行走,2 岁可跑。家属诉患儿6 岁左右出现肢体乏力,跑步姿势异常,缓慢进展,渐出现爬楼缓慢,病程中无肌阵挛发作等癫痫性发作,患儿自起病以来,智能基本正常。家族中其姐姐有类似病史,余成员均无类似病史。体格检查示下蹲起立缓慢,上臂上举缓慢,膝反射减弱,Gower征可疑阳性,巴氏征阴性,无腓肠肌肥大,无高弓足,无关节挛缩,无脊柱侧弯,无肌萎缩。辅助检测:肌电图示广泛神经源性损害,可见到纤颤电位和束颤电位等失神经电位,运动单位电位电压高,时限宽,提示脊髓前角细胞病变;脑电图正常;头部MRI正常;心肌酶谱、电解质、肝肾功能、血乳酸、血氨及同型半胱氨酸均正常。
1.2.2 先证者姐 女,13 岁9 月,第1 胎,第1 产,母孕期及出生史无异常,生长发育里程碑基本正常:1 岁2月能独立行走,2岁可跑。患儿8岁左右出现行走姿势异常,缓慢进展,渐出现爬楼困难,跑跳困难,病程中无肌阵挛发作等癫痫性发作,患儿自起病以来,智能正常。体格检查示下蹲起立困难,上臂不能完全上举,膝反射减弱,Gower征阳性,巴氏征阴性,无腓肠肌肥大,无高弓足,无关节挛缩,脊柱轻度侧弯,无明显肌萎缩。辅助检测:肌电图示广泛神经源性损害,可见到纤颤电位和束颤电位等失神经电位,运动单位电位电压高,时限宽,提示脊髓前角细胞病变;心肌酶谱示肌酸激酶(creatine kinase,CK)轻度增高;脑电图正常;头部MRI正常;电解质、肝肾功能、血乳酸、血氨及同型半胱氨酸均正常。
1.2.3 基因突变分析 经患儿父母充分知情同意后,通过安捷伦外显子芯片捕获+高通量测序行全外显子组基因测序,发现患儿携带ASAH1 基因复合杂合突变:第13内含子c.1098+1G>T杂合突变,为剪接突变,此位点为国际上已报道的致病性突变,见图1;第3 内含子c.216+11A>G 杂合突变,此位点为国际上尚未报道的新突变,国内首次报道,见图2;基因突变预测分析为可能致病变异,结合患儿临床表型,临床上考虑为致病性突变。父母来源验证显示,其父携带c.216+11A>G 突变,其母携带c.1098+1G>T 突变。线粒体DNA 高敏感性测序检测未发现异常。SMN 基因测序检测未发现异常。先证者姐姐基因测序发现携带ASAH1 基因相同位点复合杂合突变。进一步对患儿爷爷、奶奶、外公、外婆进行Sanger一代测序,结果显示均未携带相同突变,因此考虑患儿父亲,患儿母亲均为新生突变,家系图见图3。
1.2 方法

图1 患儿及家属基因测序图(第13内含子)
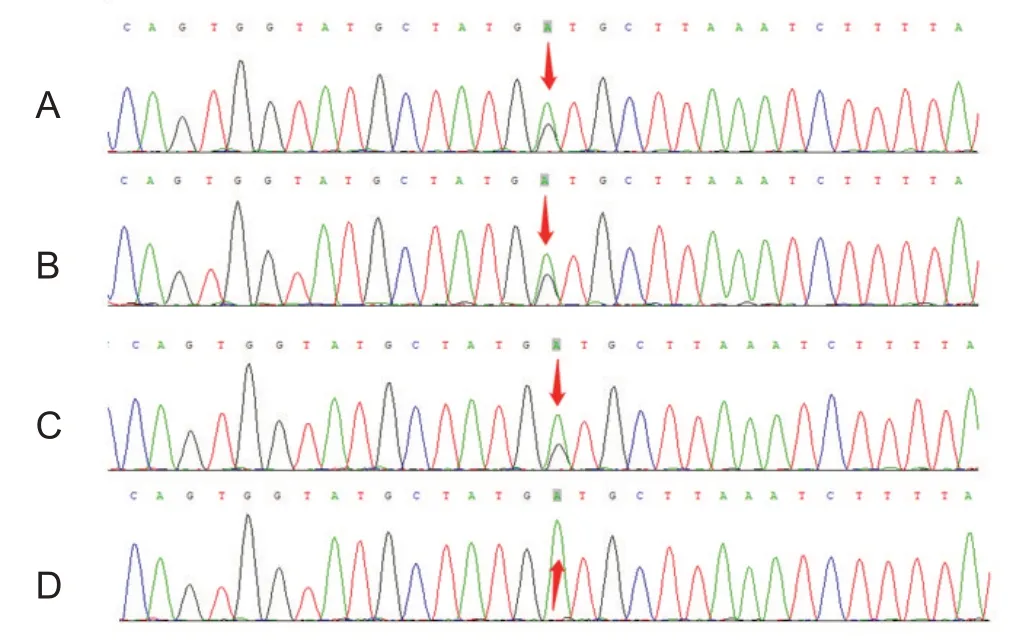
图2 患儿及家属基因测序图(第3内含子)
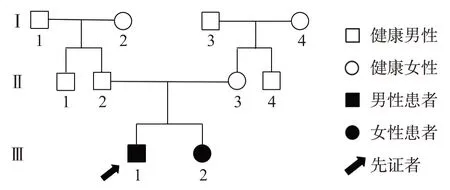
图3 ASAH1基因突变致SMA患儿家系图
收集资料并分析。在中国知网、万方数据库分别以“脊髓性肌萎缩症”和“ASAH1”进行建库至2019 年9 月文献检索;以“SMA”、“spinal muscular atrophy”和“ASAH1”检索PubMed 数据库建库至2019年9月相关文献,得到所有关于SMA的相关病例报道。采用描述性统计对符合条件的所有病例从临床表现、影像学表现、肌电图表现、实验室检查、基因测序等结果进行分析。
2 结果
检索到符合条件中文文献1篇,共1例ASAH1相关SMA 患者,其临床表型为SMA-PME。检索到英文文献8篇[2-10],报道14例ASAH1相关性SMA患者,其中12 例临床表型为SMA-PME,2 例仅为SMA,而不伴有进行性肌阵挛癫痫。14 例患者中,男性1 例,女性13例;其中以肌无力起病13 例,以癫痫发作起病1 例,以肌无力起病者,最早起病年龄为1.2岁,最晚7岁起病;12例患者病程中出现了进行性肌阵挛癫痫,癫痫发作出现年龄最早者3.8 岁,除肌阵挛发作外,还可能出现失神发作、失张力发作及强直-阵挛发作等发作形式。在病程过程中,患儿可能出现震颤、吞咽困难、反复肺炎、感音性神经性耳聋及呼吸功能不全等症状,查体可见肌萎缩、脊柱侧凸、肌张力降低、腱反射减弱及共济失调等体征。14例患儿均行肌电图检查,均提示神经源性病损;14 例患儿中10 例头部MRI 正常,4 例头部MRI提示小脑萎缩和(或)脑室扩大;11例完善了肌肉活检检查,其中9 例提示神经源性损害(失神经支配,失神经-神经再支配/神经源性肌萎缩);9例完善CK检查,8 例正常,仅1 例轻度增高;7 例完善酸性神经酰胺酶活性检测,6 例酶活性为正常细胞的10%以上,1 例<10%。所有病例均进行了ASAH1基因突变检测,共发现9种不同的突变位点,分别为:c.125C>T,c.850G>T, c.456A>C, c.886C>T, c.223insC, c.177C>G,c.256-257insA,c.124A>G及c.173C>T,其中c.125C>T最常见;所有病例均符合常染色体隐性遗传,其中6例为复合杂合遗传。在ASAH1 相关性SMA 患者中,表型为SMA-PME 者预后较差,文献报道时,已有4 例患者死亡(13~19 岁),2 例依靠机械通气及胃造口进食维持生命,而表型仅为SMA 者预后相对较好,文献报道中2例患者均存活,其中年长者已30岁。
3 讨论
酸性神经酰胺酶是一种溶酶体酶,由非糖基化的α亚基和糖基化的β亚基组成,催化神经酰胺降解为鞘氨醇和溶酶体内的游离脂肪酸,由定位在8p22的ASAH1基因编码[11]。ASAH1 基因突变相关性SMA,常见为SMA-PME表型,少数病程中仅脊髓性肌萎缩症症状,而不出现进行性肌阵挛癫痫。
SMA-PME 表型患儿症状重,起病年龄小,大约5岁左右出现肌无力症状,呈对称性,下肢重于上肢,近端肌肉受累为主,常表现为步态不稳、爬楼困难、下蹲起立困难及频繁跌倒等,肌无力症状出现数年后出现进行性肌阵挛癫痫,一般出现在7~12岁;癫痫性发作可有多种发作类型,其中肌阵挛和失神发作最多见,也可出现如眼睑肌阵挛、失神发作、失张力发作、眼睑肌阵挛持续状态等类型。此外,病程中还常出现震颤、感音神经性耳聋及脊柱侧凸等表现。患儿病情进行性加重,后期常完全丧失运动功能,累及呼吸肌可导致复发性肺部感染和呼吸衰竭,导致青少年时期死亡[8,12,13]。ASAH1基因突变仅引起SMA而不出现进行性肌阵挛癫痫者,症状较轻,进展缓慢,病程中可有震颤及脊柱侧凸等表现。此表型患儿2016年由Filosto等[10]首次报道。
本研究中2 例患儿均为学龄期起病,均以肌无力为首发症状,两姐弟(其中姐姐为病程第6 年)至今均未出现癫痫性发作,目前仅有SMA 表型,不排除病程尚短有关,尚需动态观察患儿病程中有无癫痫发作。本病神经系统检查中震颤,尤其舌肌震颤发生率较高,部分患儿可出现脊椎侧凸及肌萎缩。本研究中,姐姐有轻度脊柱侧弯,暂无震颤及肌萎缩症状,弟弟暂无相关临床表现。另外,本病实验室检查CK多数正常或仅轻度升高。本研究中姐姐CK 轻度增高,弟弟CK 正常,与之相符。本病头部MRI多正常或出现非特异性改变,如小脑萎缩或脑室扩大。本研究中两姐弟头部MRI 均正常。本病肌电图提示神经源性损伤,肌活检可表现为神经源性损伤或肌萎缩。本研究对象未行肌活检,肌电图均提示神经源性损伤。本病患者早期生长发育里程碑多正常,起病后运动功能进行性损害,而认知功能可正常或出现程度不等的损害。本研究对象早期生长发育正常,运动功能受累,认知功能正常。皮肤成纤维细胞神经酰胺酶活性的检测,可为本病的诊断提供依据,但该检测国内尚未开展。本病的确诊,除典型临床症状外,目前尚有赖于ASAH1 基因的检测,ASAH1 基因突变致酸性神经酰胺酶缺乏以致神经酰胺不能正常降解,导致神经酰胺在神经元内广泛沉积,导致相应的临床症状,但确切发病机制目前尚不清楚[10]。目前该病暂无特效治疗方法,但其等位基因病Farber 病目前已开展基因与酶替代疗法,这将为ASAH1基因突变相关SMA患者开创出未来治疗的思路[14,15]。
在临床工作中,运动障碍类疾病并不少见,单瘫性疾病需警惕良性单侧下肢肌萎缩[16],脊髓灰质炎等疾病,而当学龄前期及学龄期患儿出现肢体对称性迟缓性瘫痪,肌电图提示神经源性损伤,伴或不伴癫痫发作,则应考虑ASAH1 基因突变相关性脊髓性肌萎缩症可能,及时安排ASAH1基因检测将有助于本病的诊断。
