基于FDA“动物法则”外推超常规抗毒药物人用剂量的方法解读和思考
2020-10-28张文鹏张志伟庄笑梅
张文鹏,张志伟,庄笑梅
(军事科学院军事医学研究院毒物药物研究所,北京 100850)
自2019年12月起,新型冠状病毒(SARS-CoV-2)引发的新型冠状病毒肺炎(COVID-19)在全球迅速传播,给全世界人民生命健康带来巨大威胁[1]。在对抗新冠病毒的战役中,最为棘手的就是缺乏特效药[2]。如何在应对新发传染病时,快速提供特效药物一直以来都是全世界面临的重要课题。除了对抗突发感染,用于治疗和预防由化学物质、微生物、放射性物质等引起的严重威胁生命健康的特殊药物(包括化学药物、生物制品以及疫苗等),由于开展临床药效研究不符合伦理或不可行,研究者和审评机构需要特殊的方法和策略。基于此,美国食品药品监督管理局(Food and Drug Administration,FDA)率先颁布了“动物法则(Animal Rule)”,提出可依据适当的动物有效性实验获得的数据支持该类药物上市或储备。本文通过回顾总结目前国际公认的“动物法则”以及基于“动物法则”获批的药物,着重解读基于动物药代动力学/药效动力学(pharmacokinetics/pharmacodynamics,PK/PD)数据获得安全有效的人用剂量的原理和关键技术要点,为加快我国应急防控所需的超常规药物研发提供借鉴。
1 针对超常规药物研发国际通用的“动物法则”
针对超常规药物在开展临床有效性研究(Ⅱ期和Ⅲ期临床实验)时,因人体要接触到致死性或导致永久性损伤的毒物或微生物,不符合伦理要求而无法进行的难题,2002年7月美国FDA的药物审评与研究中心(Center for Drug Evaluation and Research,CDER)和生物制品审评与研究中心(Center for Biologics Evaluation and Research,CBER)首次颁布“动物法则”,针对化学药物的21 CFR 314.600和针对生物制品药物的21CFR 601.90[3];于2015年10月进行了修订,并以“动物原则指导下药品开发指南”发布;鉴于“动物法则”研究中方案设计结果处理等难点,2017年12月FDA专门补充发布了“基于动物法则设计研究方案与FDA沟通指南”;2019年4月再次发布了在“动物法则”指导下进行非临床研究实验室合规检查内容”。FDA如此密集的法规发布足以说明“动物法则”在超常规特殊药物审批中的重要性和紧迫性。
受到FDA“动物法则”的影响,2004年欧盟药品管理局(European Medicines Agency,EMA)发布了针对无法开展临床药效研究特殊药物的研究法规和指南[4]。2014年5月加拿大卫生部(Health Canada)也发布了特殊用途药物研发路径,其中的核心原则也是针对由化学物质、微生物、放射性物质等引起的严重威胁生命健康的特殊药物,可基于“动物法则”研发。
“动物法则”在国内已经受到关注[5],但作为中国新药审批机构的国家药品监督管理局药物审批中心,目前还没有官方发布基于“动物法则”支持超常规药物研发的指南,也没有申报和评审的案例和经验。
2 FDA基于“动物法则”批准的新药及代表性人用剂量外推案例
基于“动物法则”研究的药物,由于无法开展大规模人体药效研究(如有限临床病例研究结果,作为重要数据补充),非临床的研究结果更加重要。因此,“动物法则”特别强调要包含以下4个标准(表1),才能保证获得的外推人用剂量是可信的。同时,必须有Ⅰ期临床研究数据,以获得药物在健康人体中的剂量-暴露(PK)特征,以及安全剂量范围的数据。
到目前为止,FDA基于“动物法则”共审批了14个超常规药物用于军事储备或应对突发公共卫生事件,包括针对化学物质、微生物、放射性物质的化药、生物药以及疫苗等[6]。表2按照FDA批准的时间顺序总结了14个超常规药物的主要信息,包括必须符合的4个研究标准以及人用剂量桥接策略。从可获得的信息可以看出,这些药物分为增加新适应症的“老药新用”,以及全新的药物或疫苗两类。对于增加新适应症的已上市药物来说,由于已有完整的临床前研究数据和针对原适应症的临床研究数据(有时还包括临床超标签应用的报道),可以大大缩短研发周期。对这类药物,不必重复进行临床前研究及Ⅰ期临床研究,而是根据“动物法则”4个研究标准,建立新适应症的动物模型及体外模型。所采用的动物模型的发病机制,临床表现,以及终点指标都要与人体具有高度可比性。在这种情况下,获得该模型动物PK/PD,根据人体药代参数(药物暴露)及药效相关依据,合理外推患者体内暴露及剂量,替代临床Ⅱ期或Ⅲ期研究。此类药物包括抗菌素左氧氟沙星,盐酸环丙沙星用于治疗炭疽感染,非格司亭、培非格司亭、沙格司亭用于治疗辐射损伤等。对于全新的药物或疫苗来说,按照“动物法则”研究和申报超常规药物,必须先进行系统的临床前药代动力学、安全性评价以及临床Ⅰ期研究,然后按照4个研究标准进行人体药效剂量的外推。这类药物包括美军研发的预防梭曼中毒药物溴吡斯的明、治疗炭疽感染的雷昔库单抗、奥比妥昔单抗,以及抗天花的特考韦瑞等。

Tab 1 Four criteria for FDA to rely on evidence from studies in animals to provide substantial evidence of effectiveness of these products
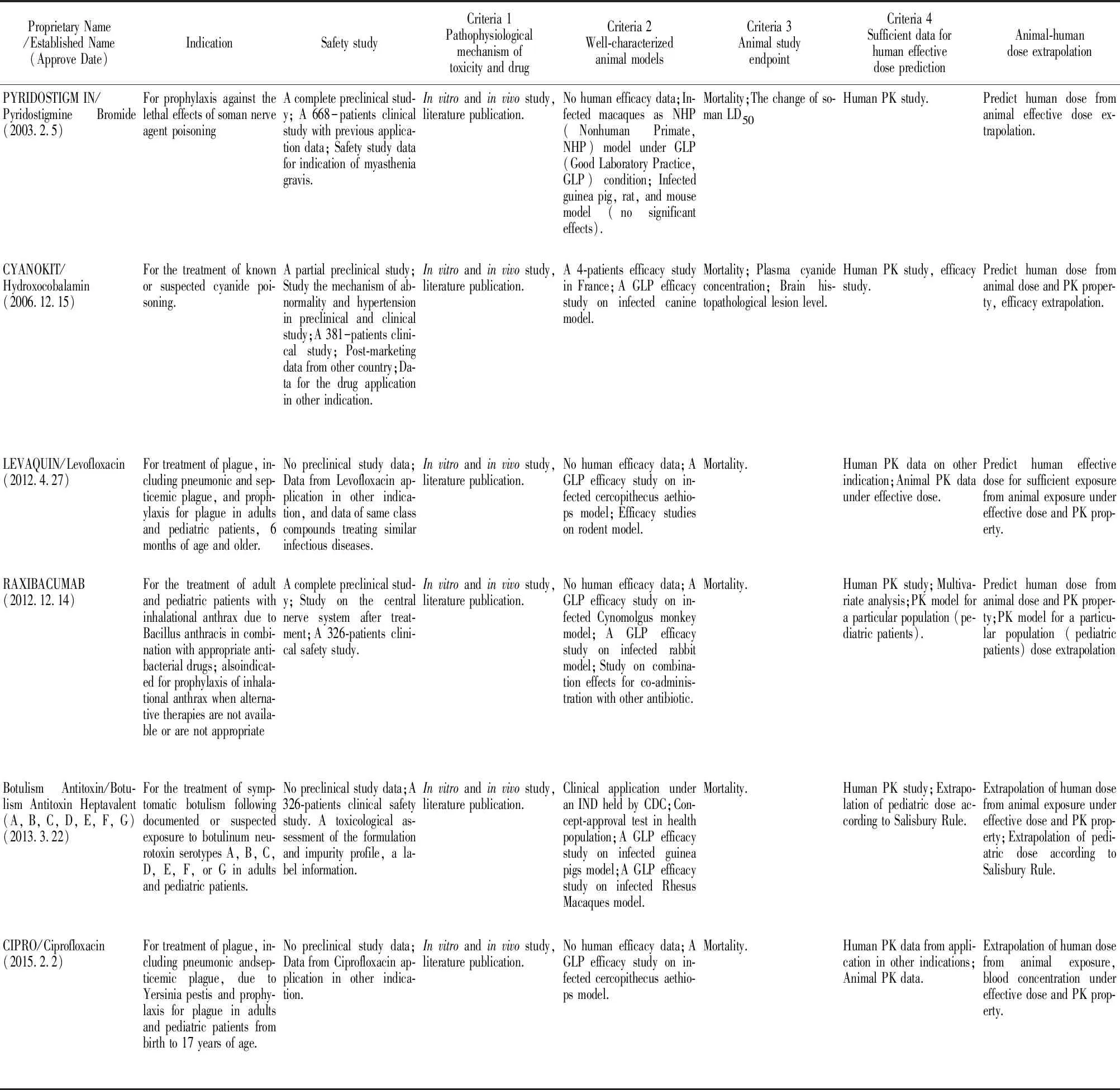
Tab 2 Summary of FDA approved drugs under “Animal Rule”
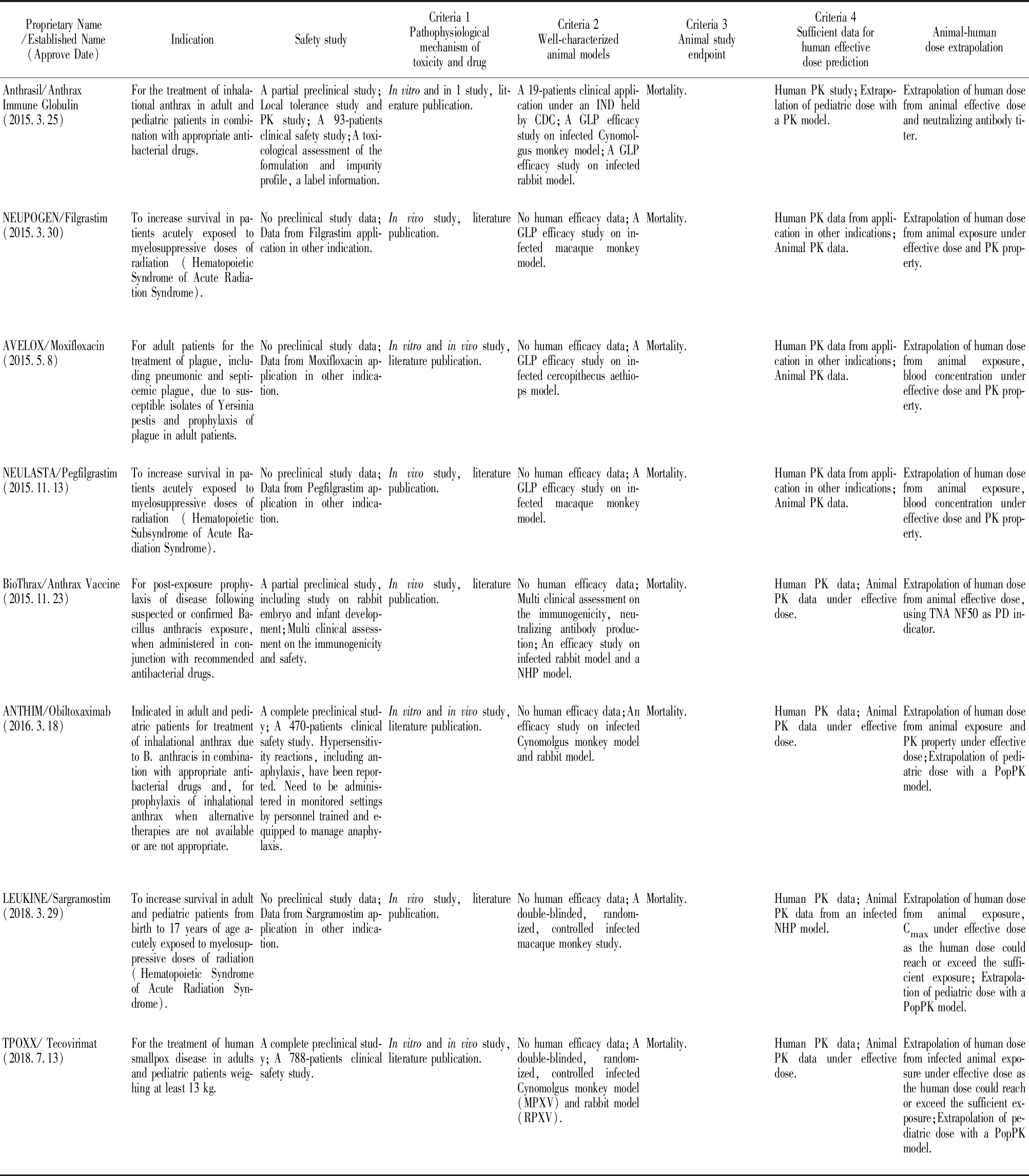
Tab 2
本文以特考韦瑞和奥比妥昔单抗为例,简要阐述如何依据临床前研究、动物有效数据以及临床Ⅰ期研究结果外推全新结构的小分子化药和大分子单抗药物的临床有效性,并外推人用剂量。
特考韦瑞(Tecovirimat,TPOXX,又名ST-246)是FDA依据“动物法则”最新批准的首个抗天花病毒全新化学药物,整个研发过程非常值得借鉴。天花病毒作为烈性病毒,受到伦理限制,无法进行临床有效性研究,必须依据“动物法则”外推人体有效剂量[7]。天花病毒是痘病毒(Orthopoxvirus)的一种,仅感染人类,出现皮肤发疹和发热,以至死亡,因此,按照动物法则要求,建立合适的动物模型具有一定的挑战性。最终,研究人员利用药物靶点在痘病毒中的保守性,采用猴痘病毒(monkeypox virus,MPXV)与兔痘病毒(rabbitpox virus,RPXV)两种不同的强致死性痘病毒作为替代,建立了食蟹猴与兔两种染毒模型[8],并证明这两个动物模型中均可出现与人类相似的病毒血症、皮肤痘疹和高死亡率,成功完成了动物模型构建。TPOXX抗天花病毒靶点是病毒复制必须的包封蛋白,这与其在动物体内发挥抗痘病毒作用是一致的。因此TPOXX在动物模型中获得的药代和药效(存活率)数据能够可信地桥接于人体。经过临床前以及动物模型中的PK/PD研究,证明了TPOXX对病毒的有效性(兔模型40 mg·kg-1·d-1,猴模型10 mg·kg-1·d-1,连续14 d给药有效率可达95.5%[14]和96%[10])及其药物暴露的药代参数(药时曲线下面积(area under curve,AUC),峰浓度Cmax和谷浓度Cmin)。通过比较TPOXX健康人体(共449例,其中359例接受了600 mg,每天两次(Bis in die,BID),口服剂量)研究发现,连续14 d餐后口服600 mg BID,能够在人体内获得超出动物有效剂量水平的暴露量(表3[9-10]),并且是安全的。同时,能否将药物的正常人体PK数据基于“动物法则”外推到感染患者,还需证明病毒感染是否影响药物在体内的PK暴露特征,以及影响药物在动物和人体PK特征的因素是否存在明显物种差异。在这个案例中,研究者应用群体药动模型(Population Pharmacokinetics,PopPK),首先证明了正常动物与病毒感染动物PK特征无明显差别,并进一步证明了(影响药物在动物和人体PK特征的因素无明显的种属差异[11]。最终,TPOXX的人用剂量被设定为餐后口服600mg BID,连续给药14 d。在缺乏临床有效性研究的情况下,TPOXX应用动物-人体在PK/PD的桥接获得了FDA的认可。
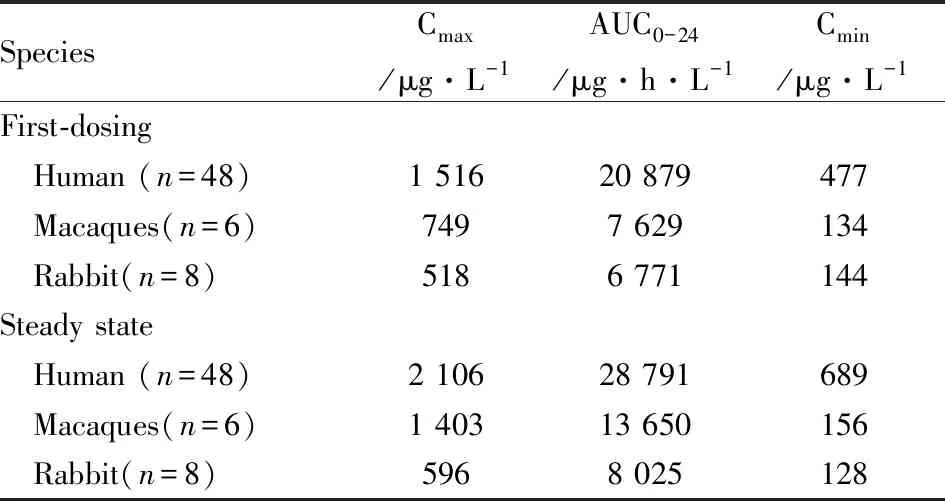
Tab 3 PK parameters of TPOXX in uninfected human, MPXV infected macaques and RPXV infected rabbits [9-10]
奥比妥昔单抗(ANTHIM)是FDA继批准瑞西巴库单抗以后,依据“动物法则”批准的第二个治疗和预防炭疽感染的单抗。机制研究证明,ANTHIM可以特异性中和炭疽毒素-保护性抗原(protective antigen,PA),后者是炭疽杆菌造成机体损伤的毒素蛋白。新西兰兔和食蟹猴吸入炭疽孢子后建立的动物模型与人体的发病机制和特点一致[12],因此用于动物有效性研究。兔和猴吸入感染200×LD50气溶胶态炭疽杆菌后,应用ANTHIM(静注剂量为1~32 mg·kg-1,并包括安慰剂对照组)对抗,实验终点指标是28 d存活率,并测定血清中ANTHIM和PA含量。动物模型表明ANTHIM 16 mg·kg-1能明显提高兔和猴生存率(超过ED90的概率80%),并达到药效平台[13]。同时进行联合左氧氟沙星实验,发现合用抗生素未明显提高生存率。在临床Ⅰ期超过500例健康受试者进行了4~24 mg·kg-1剂量范围的安全性和PK研究,还包括了合用抗生素的PK研究。比较ANTHIM的在染毒后兔和猴的PK以及健康人体PK,发现同样静注16 mg·kg-1后人体中药物峰浓度(Cmax)与动物模型相当,但AUC几乎是动物的两倍(图2,表4)[14],合用抗生素不改变ANTHIM 的PK。由于抗体药物的体内清除具有明显的种属差异,人体的半衰期明显高于动物,因此人体内暴露量高于动物[15]。考虑到靶点介导的清除(Target Mediated Drug Disposition,TMDD)是抗体药物的另一个重要清除机制,而在健康人体中缺乏PA这一特异靶点,研究人员应用PopPK进行了建模和模拟。模型结果显示,预测的Cmax和AUC在健康人体体内与实测结果吻合度良好,而感染后患者体内的AUC比健康人体降低了18%(表4),主要是由于TMDD造成的,但是这并不会降低药物的抗毒作用。因此,最终确定16 mg·kg-1为成人患者用量。在PopPK建模过程中,通过考察年龄、性别、种族等各种协变量对PK/PD的影响,发现体质量是个敏感参数。因此,为了给儿童患者提供有效剂量,将体质量作为协变量,获得了不同体质量儿童的AUC和Cmax,计算出儿童给药剂量为15 kg以下32 mg·kg-1,15~40 kg按24 mg·kg-1给药。
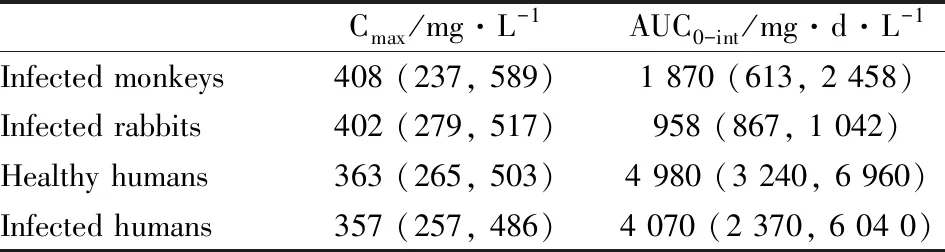
Tab 4 Overview of pharmacokinetic analyses for i.v. dose of 16 mg·kg-1 ANTHIM in animal models and humans[14]
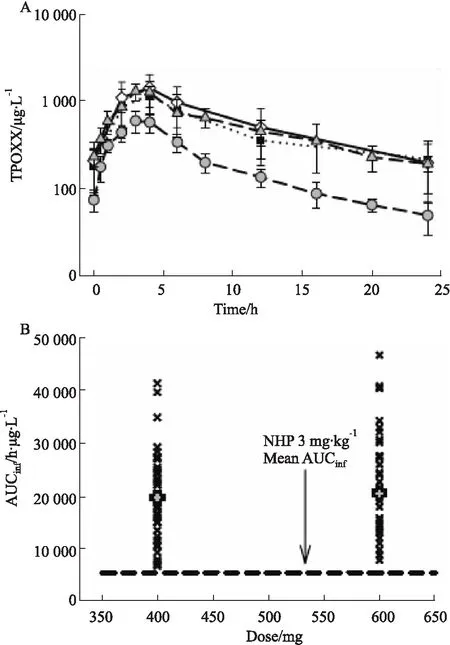
Fig 1 Plasma concentration-time curves (A) and individual exposure levels (AUC, B) for uninfected humans and NHPs over a range of TPOXX doses relevant to efficacious-dose.
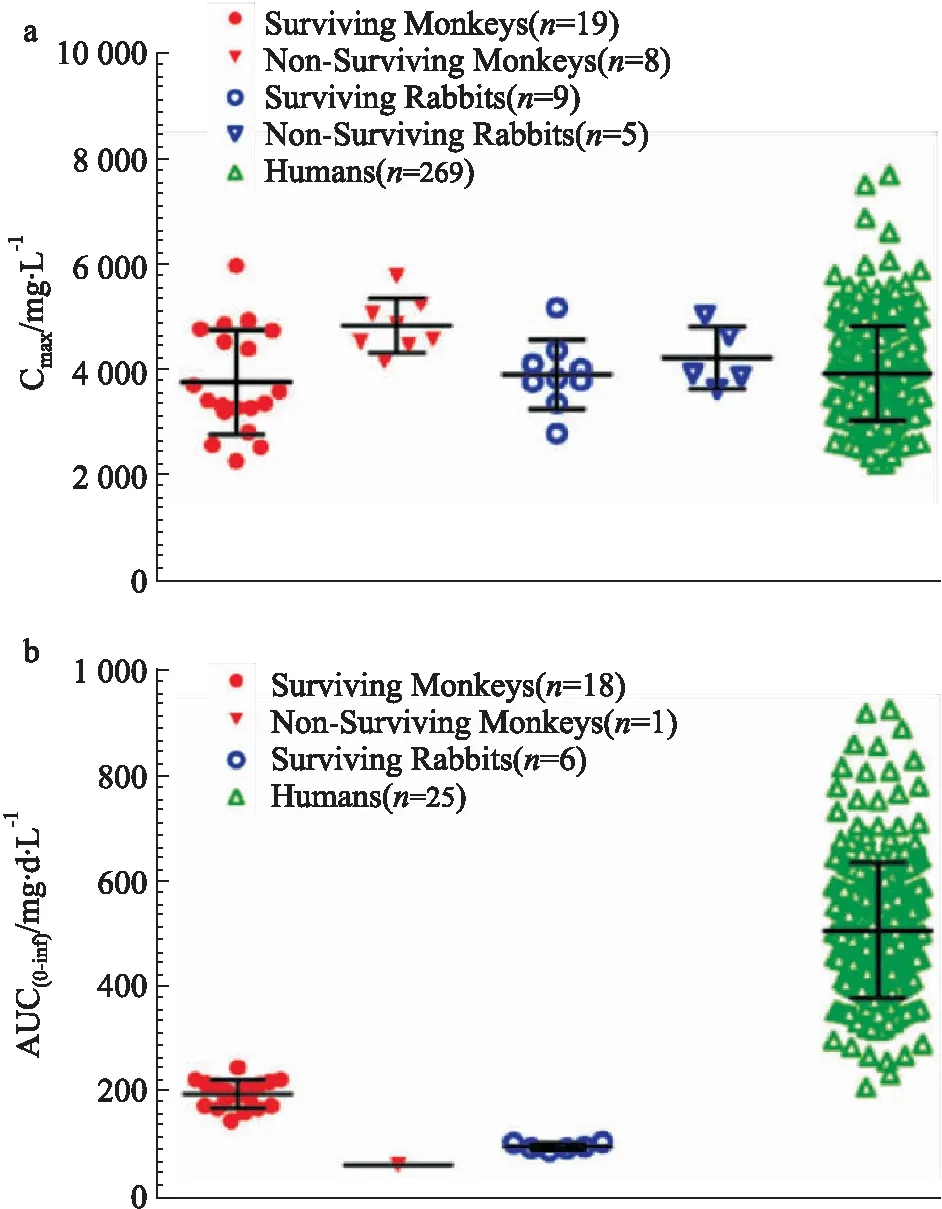
Fig 2 ANTHIM exposure in animal models and humans after i.v. dose of 16 mg·kg-1 (a) Cmax, (b) AUC [14]
3 超常规抗毒药动物剂量外推人用剂量的关键技术要点
“动物法则”用于指导无法开展临床有效研究但具有重要需求的特殊药物,它制定的4个基本标准比一般药物研发的临床前研究标准严格得多,目的就是保障动物有效剂量的人化外推是安全有效的。鉴于人化剂量的获得是“动物法则”中的核心步骤,比较体内暴露是桥接动物和人的关键环节。结合动物法则指南要求、已获批特殊药物的研究以及作者的实践工作体会,总结主要关键技术要点如下。
3.1 PK/PD(药代动力学/药效动力学)虽然PK研究是获得药物体内暴露特征的关键,但在正常动物体内进行的PK由于忽略了病理状态对药物体内处置的影响,不能作为外推人体剂量的唯一参考。只有在动物疾病模型中获得的PK,同时结合PD指标,才是外推人体的重要依据。因此,动物模型中PK/PD关联是动物有效剂量外推至人的最重要方法。具体应用时,针对不同药物可采取不同策略。
(1)如果药物的抗毒机制是直接对抗外源毒物而不是作用于机体本身,体外模型上获得的有效暴露浓度可用于估算体内暴露所需的有效浓度。实际应用时,可以首先在动物模型上进行PK/PD验证,再根据人体Ⅰ期临床获得的PK结果,反推人体得到相应PD指标所需的人化剂量。基于这一思路,抗菌素基于PK/PD人用剂量的预测比较准确。反之,如果药物的作用机制或靶点是机体本身,通过改变机体细胞蛋白的内在功能而抵御感染或外源毒物,应用动物模型PK/PD外推人体时可能因存在宿主特异性的障碍,使得依据“动物法则”进行有效剂量外推的可靠性大打折扣。目前,FDA按照“动物法则”批准的抗毒药物中,其作用靶点都是直接针对病毒或细菌的。
(2)如果受试药已被批准用于其他适应症,参考已有的人体PK/PD数据,在新的动物模型中获得的PK/PD可进行针对新适应症的人用剂量转换,也是非常可行的。
(3)如果药物的临床药效基于合适的生物标志物,则必须证明这个生物标志物与产生临床期望疗效相关联,并且在动物模型中必须能够检测到相同的生物标志物,这时动物的PK/PD也可以转化为人体的PK/PD。其中必须注意的是,在没有足够证据证明动物和人体的反应(生物标志物,MIC等)一致或相关时,动物体内获得的PK/PD不能直接外推到人。在这种情况下,我们只能假定建立的动物E/R(暴露/反应)关系与人是类似的,并应用保守的方法进行动物到人体的外推。同时,由于没有替代指标,可以根据药物的暴露参数(AUC、Cmax、Css)确定人用剂量。
(4)需要确定产生药效的是药物原型还是其代谢产物,如果是代谢产物,必须建立活性代谢产物的PK/PD,同时要证明代谢转化模式在动物模型和人是一致的。
3.2 物种差异由于药物的体内吸收、分布、代谢、排泄(ADME)等关键因素存在物种差异,影响药物浓度-时间曲线,在进行动物剂量人化外推时必须根据种属差异适当调整。
(1)为了充分获得PK物种差异,至少要在动物模型中进行3个剂量的PK研究,以绘制出完整的暴露-效应曲线,获得最大药效剂量和动物模型体内的PK特征。
(2)药物ADME的物种差异比较可在体外模型上进行。其中,人和动物肝细胞或肝微粒体模型能够较快速地准确获得小分子药物经肝脏代谢的种属差异。
(3)药物蛋白结合率的物种差异也是非常重要的影响因素,但常常被忽略。由于只有游离的药物才能够发挥药效,在进行PK/PD的体内外和种属桥接转化时,必须根据血浆,以及组织、细胞的蛋白结合率校正后[16],按照游离药物浓度进行,否则可能会导致严重偏差。
(4)充分获得药物在模型动物和人的ADME物种差异后,可通过调整动物的给药剂量和方式,使血药浓度-时间曲线更接近人体,获得精准的人化剂量。随着模型介导新药研发(model based drug development,MBDD)模式的发展,基于生理药动学(physiologically based pharmacokinetic,PBPK)模型和群体药动学(PopPK)模型也为人用剂量的获得提供了更好的技术平台,同时还为特殊人群(儿童、老人等)用药剂量的调整提供了可能[17]。
3.3 药物相互作用对于致命性毒物、微生物等造成的损伤,临床救治时除了抗毒药以外,通常会合并使用多种药物。药物联合应用时由于受到基于ADME的药物-药物相互作用影响,常常会发生药代动力学性质的改变,影响药效和安全性。因此在动物剂量人化时也需根据受试药的代谢特征进行必要的药物相互作用研究。药物-药物相互作用(Drug-drug Interaction,DDI)可以在体外模型或健康人体内进行,具体可参考FDA发布的DDI指南[18]。
综上,“动物法则”对于无法开展临床有效性研究的超常规抗毒药的研发具有显著的科学指导意义。尽管无法开展临床有效性研究,如果严格满足FDA“动物法则”的基本标准,将有足够的信心根据动物实验结果外推获得准确的人用有效剂量,解决这类药物研发的瓶颈。在当前形势下,我国药品监管机构需要尽快制订符合我国医疗实践和国情的“动物法则”指南。国内药物研究机构也需着眼这一重大需求,建立相关技术平台,多学科密切协作,提高国家生物安全防御能力。
