Microbial Community Composition and Function in Sediments from the Pearl River Mouth Basin
2020-09-27CHENYeMITiezhuLIUYuetengLISiqiandZHENYu
CHEN Ye, MI Tiezhu, LIU Yueteng, LI Siqi, and ZHEN Yu, *
Microbial Community Composition and Function in Sediments from the Pearl River Mouth Basin
CHEN Ye1), 2), 3), MI Tiezhu2), 3), 4), LIU Yueteng3), 4), LI Siqi1), 2), 3), and ZHEN Yu2), 3), 4), *
1)College of Marine Life Sciences, Ocean University of China, Qingdao 266003,China 2) Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266071, China 3) Key Laboratory of Marine Environment and Ecology, Ministry of Education, Qingdao 266100, China 4) College of Environmental Science and Engineering, Ocean University of China, Qingdao 266100, China
This study was conducted to characterize the diversity and function of microbial communities in marine sediments of the Pearl River Mouth Basin (PRMB) in the South China Sea. The results showed that the bacterial and archaeal communities varied greatly with depth. Proteobacteria in bacterial communities and Nitrososphaeria and Woesearchaeota in archaeal communities were dominant in the shallow sediments (1-40cm), while Chloroflexi in bacterial communities and Bathyarchaeia in archaeal communities were dominant in the deep sediments (50-200cm). Regarding ecological functions based on the metatranscriptomic data, genes involved in various pathways of nitrogen metabolism and sulfur metabolism were observed in the tested sediment samples. Metagenomic analysis revealed that Proteobacteria contribute the most to nearly all genes involved in nitrogen and sulfur metabolism. Moreover, Thaumarchaeota contribute the most to certain genes involved in nitrification, denitrification and assimilatory sulfate reduction pathways. The most abundant bacterial genus,is crucial for nitrification, dissimilatory nitrate reduction, denitrification and assimilatory sulfate reduction pathways.
microbial community and function; high-throughput sequencing; Pearl River Mouth Basin (PRMB)
1 Introduction
Prokaryotes, which include the two domains bacteria and archaea, play important roles in the transformation of organic matter and the cycling of biogeochemical elements in marine sediment. Bacterial communities in marine sediments typically consist of a number of ubiquitous phyla, including Proteobacteria, Chloroflexi, Planctomycetes and Acidobacteria (Inagaki., 2006; Nunoura., 2012; Liu., 2015; Mahmoudi., 2015). Based on phylogenetic surveys, among these bacteria, Proteobacteria are predominant in marine sediments, and associated with biogeochemical cycles such as the carbon, nitrogen, or sulfur cycles (McCaig., 1999; Lenk., 2011; Varon-Lopez., 2014). The phylum Chloroflexi was reported to be dominant in deep subsurface sediments, as it could occupy up to 70% of the bacterial 16S rRNA sequences in the Mediterranean sediment (Coolen., 2002). Different subgroups of Chloroflexi are suggested to have ecological functions, such as CO2fixation, aromatic and fatty acid oxidization, acetate production and dimethyl sulfoxide utilization (Hug., 2013; Was-mund., 2014). Archaea are commonly found in marine sediments, with Thaumarchaeota often dominating the microbial community in marine sediments (Inagaki., 2003, 2006; Nunoura., 2012; Liu., 2015). Thaumarchaeota include almost all aerobic ammonia- oxidizers, suggesting that they might play important roles in biogeochemical nitrogen cycling (Könneke., 2005; Brochier-Armanet., 2008). Moreover, some cases, for example, Bathyarchaeia within Crenarchaeota, can constitute up to 71% of the microorganisms in volcanic ash layers in the Okhotsk Sea, as revealed by 16S rRNA gene sequence analysis (Coolen., 2002; Inagaki., 2003).
The South China Sea (SCS) is one of the largest marginal seas around the Western Pacific Ocean, covering a total area of 3.5millionkm2with an average depth of over 2000m (Xie., 2003). Previous studies have focused on the diversity and the functional roles of microbial communities in various areas of the SCS (Zhang., 2012, 2014; Jiao., 2015; Liu., 2015; Gong., 2017). For instance, in the Shenhu area, microbial communities were influenced by hydrate and geochemical variable, and could be differentiated from other hydrate- related sediments around the world (Jiao., 2015). And in the Taixinan Basin, the dominant bacterial/ar- chaeal groups observed were mainly involved in organic matter decomposition, sulfur-oxidation and methane generation (Gong., 2017). In the cold seep of the northern South China Sea, the microbial structures revealed a coupled reaction of sulphate reduction and methane oxidation (Zhang., 2012). All mentioned above showed that the microbes are highly diverse and play a vital role in the biogeochemical cycles in the sediments of the SCS. These results were based on an analysis of the 16S rRNA gene and functional genes. However, amplicon sequencing cannot overcome PCR in obtaining accurate qualitative and quantitative biodiversity data. In contrast, high- throughput metagenomic sequencing that obtains whole genome sequences from environmental samples can avoid PCR bias. This method can simultaneously demonstrate both the taxonomic and functional diversity of a microbial community and become a powerful tool to provide an all-inclusive picture of the functional potential of an ecosystem.
In the present study, we conducted genotyping assays via two approaches (sequencing of 16S rRNA gene amplicons and metagenomic analysis) on the Illumina HiSeq 2500 platform to analyze the microbial community structures and functional profiles in sediments from the PRMB in the northeastern SCS. Our primary objectives were i) to reveal the vertical distribution of the microbial communities and their potential environmental drivers and ii) to investigate the functional genes and microbes involved in nitrogen and sulfur biogeochemical cycles.
2 Materials and Methods
2.1 Sample Collection
Two sediment cores were obtained in the site S8 in the PRMB of the SCS during a research cruise in May 2016. The location was 19.88˚N, 115.15˚E (Fig.1). One sediment core (S8.1, length: 40cm) was collected by using a box corer, and the other sediment core (S8.2, length: 200 cm) was collected by using a gravity corer. Water depth, temperature, salinity and oxygen saturation of the bottom waters were measured using a conductivity-temperature- depth (CTD) system (Table 1). After collection, the 1-, 20- and 40-cm layers of sediment core S8.1 were sampled for microbial analysis, and the 1-, 50-, 100-, 150- and 200-cm layers of sediment core S8.2 were sampled for microbial analysis. It is worth mentioning that certain shallow marine sediments break away when using the gravity corer. Thus, we used sediment core S8.1 to make up in the analysis for the loss of shallow sediments in core S8.2.
Fig.1 Location of sampling site in the PRMB of the SCS.
2.2 Environmental Factor Analysis
Sediment pH was measured after mixing the sediments with deionized water free of CO2at a ratio (se- diment/water) of 1:5. NH4+, NO2−, and NO3−were extracted from sediments with 2molL−1KCl as previously described (Hou., 2013) and quantified with a QuAAtro nutrient auto analyzer (Seal Analytical Ltd., UK). The total organic carbon (%TOC) was measured after removing inorganic carbon by digesting with HCl and using an elemental analyzer (PE2400II, UK).

Table 1 Environmental parameters of the bottom waters
2.3 DNA Extraction
Total genomic DNA was extracted from approximately 0.25g of wet weight sediment using a PowerSoil DNA Isolation Kit (Mo Bio, USA) according to the manufacturer’s protocol.
2.4 Quantitative PCR
Quantitative real-time PCR was performed to quantify the abundance of the total bacteria and archaea in the eight core samples as previously described (He., 2015) with minor modification. The bacterial 16S rRNA gene copies were quantified using the universal primer pair 338F/806R (Peiffer., 2013), and the archaeal 16S rRNA gene copies were quantified using the primer pair U519F/806R (Porat., 2010). A 20-µL reaction for the amplification of the 16S rRNA gene contained 10 μL of FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics, Germany), 0.3μL of each primer, 0.2 μgμL−1bovine serum albumin (BSA) and 2.0μL of sediment DNA. The procedure employed for the PCR was as follows: an initial denaturation step at 95℃ for 10min and 40 cycles of 95℃ for 15s and 58℃ for 2min. The assays were performed on an ABI PRISM® 7500 Sequence Detection System (Applied Biosystems, USA) with ABI PRISM 7500 software (version 1.3.1). The standard curves for each assay were obtained by using serial 10-fold dilutions of plasmids carrying the target gene fragments. Only standard curves with linear relationships and2>0.99 were used. Amplification effici- encies were 0.90 and 0.99 for bacteria and archaea, respectively. All qPCRs were performed in triplicate for each sample.
2.5 16S rRNA Gene Pyrosequencing and Data Analysis
Total DNA from the eight core samples was used as templates for amplification using barcoded primers (515F/806R for bacteria and Arch519F/Arch915R for archaea) (Coolen., 2004; Hori., 2014) targeting the 16S ribosomal RNA (rRNA) genes. Sequencing was performed on the Illumina HiSeq 2500 PE250 platform (Total Genomics Solution Institute, Shenzhen, China). The raw data were processed using the QIIME pipeline (Caporaso., 2010). Raw reads that had a quality score higher than 20 over a 50-bp window size and a minimum length of 50 bp were retained (Bokulich., 2013). The pair-end reads were joined with at least a 10-bp overlap using FLASH (Magoč and Salzberg, 2011). After quality control, UPARSE was employed to cluster all of the clean reads into operational taxonomic units (OTUs) at a 97% dissimilarity level with default parameters. The most common sequences from each OTU were selected as the representative sequences. Then, taxonomy was assigned against the SILVA 132 databaseQIIME with a confidence level of 80%.
To fairly compare all the samples at the same sequencing depth, sequences were randomly reduced in number to the smallest read numbers in each sample. Then, diversity estimators (Chao1, Shannon and Simpson) and Good’s coverage estimator in each library were calculated using the QIIME software package. For beta diversity, the cluster dendrogram based on Bray-Curtis dissimilarity at the OTU level was constructed with the R package vegan to describe the relationship of bacterial and archaeal communities among samples. Redundancy analysis (RDA) with Monte Carlo permutation tests (999 unrestricted permutations,<0.05) was conducted using the R package to reveal the correlation between community composition and environmental factors. Spearman’s correlation analysis for microbial abundance, alpha diversity, percentage composition of taxa and environmental factors was performed by using SPSS statistics software (v17.0 for Windows).
2.6 Metagenomic Analysis
The metagenomic DNA isolated from 1- and 40-cm samples from S8.1 was sent to the Total Genomics Solution Institute (Shenzhen, China) for metagenome library construction and Illumina high-throughput sequencing. Paired-end (PE) raw reads were generated after sequencing, and clean data were extracted from the raw reads following the removal of adaptor fragments and low- quality reads. After quality control, clean reads were then assembled into scaffolds by using IDBA software (Peng., 2010). Then, open reading frames (ORFs) were predicted using MGA (version 2.10), and a BlastP search (v2.2.30) was conducted on the ORF sequences, comparing these sequences to those in the National Center for Biotechnology Information (NCBI) non-redundant protein (‘nr’) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases with an E-value cut-off of 1e−5to obtain functional and taxonomy annotation.
2.7 Nucleotide Sequence Accession Numbers
The sequence data generated in the present study have been deposited into theNCBI SRA database under the accession numbers SRP102424 (16S rRNA gene) and SRP102424 (metagenomic datasets).
3 Results
3.1 Environmental Factors at Sampling Stations
Detailed environmental factors of the sediment depth profile were shown in Table 2. In terms of inorganic nitrogen contents, higher NH4+concentrations were observ- ed in the shallow layers (S8.1:1–40cm and S8.2–1cm) than in deep layers (S8.2: 50–200cm). There was no obvious trend for NO3−and NO2−the sediment depth. Sediment pH varied from 7.62–8.37, increasing with sediment depth. The TOC content showed a clear decreasing trend with increasing sediment depth, with a range of 0.49%– 0.86%.

Table 2 Environmental parameters of samples in different sediment samples
3.2 Quantitative PCR
The results of the real-time PCR experiment showed that the abundance of bacteria and archaea ranged from 1.29×107copiesg−1to 6.93×108copiesg−1(wet weight) and 2.04×106copiesg−1to 8.20×107copiesg−1(wet weight), respectively, in the eight core samples (Fig.2). The archaeal gene copy numbers were equivalent to approxima- tely 9.18%–15.79% of the bacterial gene copy numbers at corresponding depths. Meanwhile, the abundance of the bacterial and archaeal 16S rRNA genes decreased with increasing depth, decreasing by one order of magnitude at depths greater than 50cm.
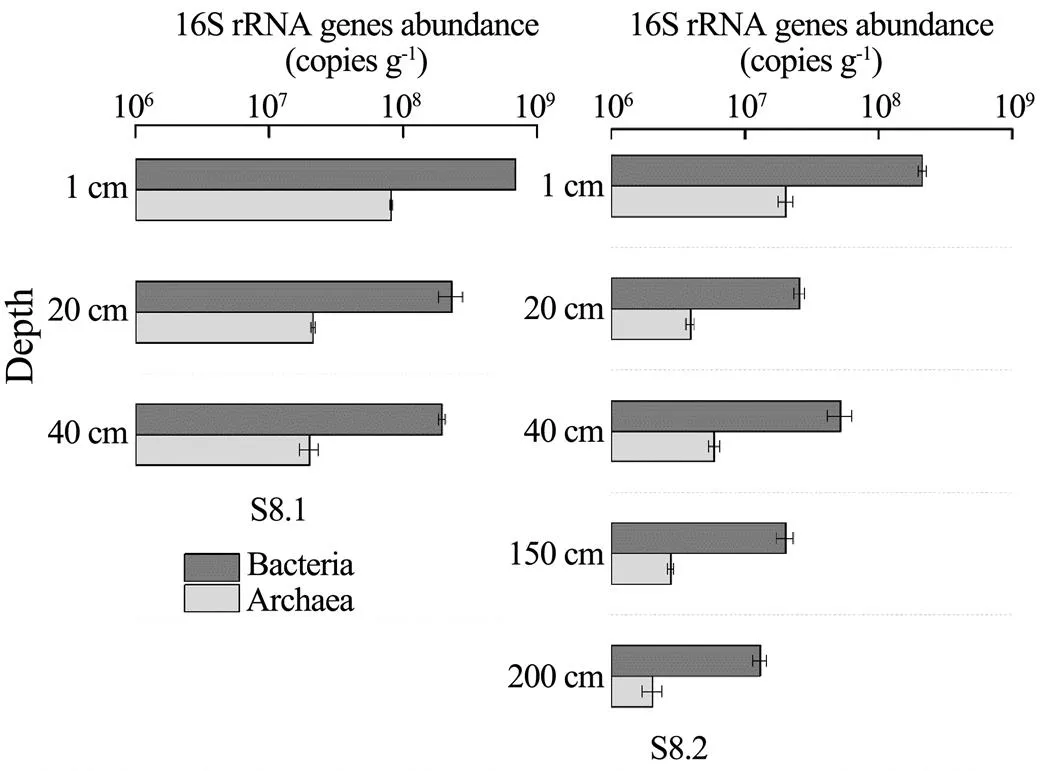
Fig.2 Bacterial and archaeal abundance in different sediment samples.
3.3 Bacterial and Archaeal 16S rDNA Analysis
Illumina-based analysis of the 16S rDNA amplicons recovered a total of 222033 high-quality bacterial sequences with an average length of 256bp from the eight core samples (Table 3). When OTUs were grouped at a 97% similarity level, there were a total of 3813 OTUs, ranging in number from 886 to 1918, in the eight core samples (Table 3). Good’s coverage estimator revealed that 96.59%–98.83% of the bacterial OTUs were present in each sediment sample, which indicated that a majority of the bacterial OTUs had been captured. The Chao1 index varied between 1083 and 2567, and the Shannon index varied between 7.29 and 8.97. These indices show- ed that both species diversity and richness were decreased in the deep sediments (50–200cm) of sample S8.2.

Table 3 The summary of sequences, OTUs, diversity index, richness index and Good’s coverage of the bacterial and archaeal communities
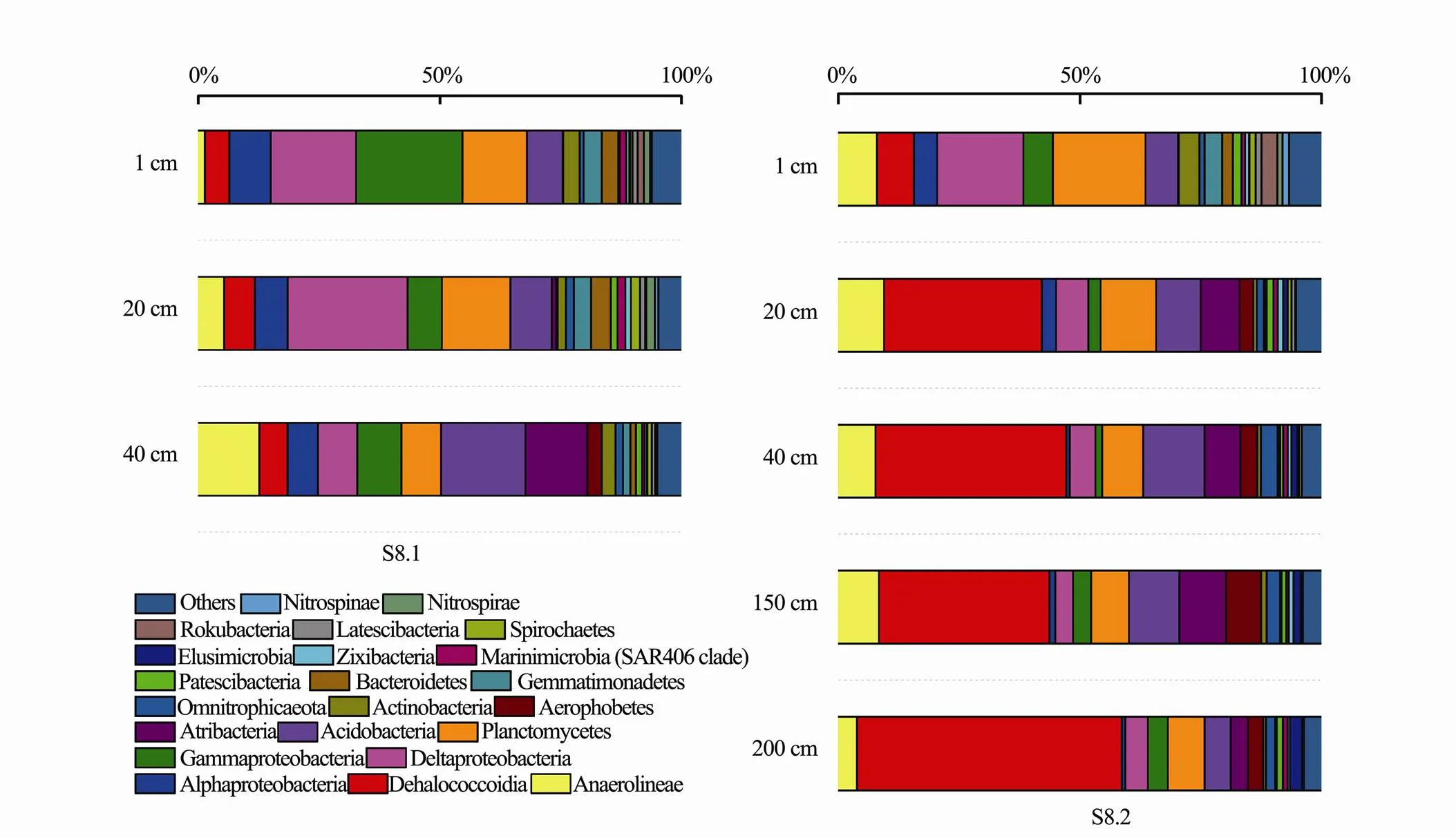
Fig.3 Relative abundance of bacterial phyla (including classes of Proteobacteria and Chloroflexi) in different sediment samples with abundance >1%.
A total of 53 bacterial phyla were identified from the eight core samples. The communities were dominated by Chloroflexi, Proteobacteria, Planctomycetes and Acidobacteria (Fig.3). In this study, we identified Chloroflexi (31.27% of the bacterial sequences) and Proteobacteria (22.12% of the bacterial sequences) as the predominant bacterial groups. Dehalococcoidia (DEH) was the most abundant class within Chloroflexi. The relative abundance of this class increased with sediment depth, making it the most dominant class in the deep layers (50-200 cm) of sample S8.2. Within Proteobacteria, the predominant group was Deltaproteobacteria (11.10%), followed by Gammaproteobacteria (6.99%) and Alphaproteobacteria (4.02%). These two groups were dominant in the shallow sediment layers (1-40cm) of S8.1 and S8.2, and their relative abundance decreased with sediment depth. Sequences belonging to Planctomycetes were detected at all sediment depths, and this phylum mainly dominated the shallow sediments.
A total of 206612 high-quality archaeal sequences with an average read length of 385 bp were obtained from the eight core samples, and each sample contained 14,988 to 31842 reads (Table 3). Each archaeal library was composed of 971 to 1315 OTUs at 97% sequence similarity. High Good’s coverage revealed that ≥96% of the archaeal OTUs were well captured in each sediment sample, indicating that sufficient sequencing depth was achieved for archaeal sequences. The Chao1 index varied between 1236 and 1893, and the Shannon index varied between 6.13 and 7.54. The Chaol and Shannon indices showed that the archaeal richness and diversity were lower than the richness and diversity of the bacterial communities.
Nine different archaeal phyla were detected across the sediment samples. The predominant phyla in the sediment samples were Crenarchaeota (38.90%) and Thaumarchaeota (21.89%), followed by Nanoarchaeota (12.87%), Asgardaeota (11.33%) and Euryarchaeota (7.91%) (Fig. 4). Within Crenarchaeota, a majority of the sequences were assigned to the class Bathyarchaeia (38.50%). The relative abundance of this class increased with sediment depth, making Bathyarchaeia the most dominant class in the deep sediment layers (50–200cm) of sample S8.2. Nitrososphaeria was the most abundant class within Thaumarchaeota, which dominated the shallow sediment layers (1–40cm) of S8.1 and S8.2 and even accounted for 75.63% of the surface sediment (1cm) of S8.1. The archaeal sequences associated with Woesearchaeota had the highest relative abundances in the shallow sediment layers (1–40cm) of samples S8.1 and S8.2.

Fig.4 Relative abundance of archaeal classes in different sediment samples with abundance >1%.
3.4 Microbial Community Comparison and Environmental Factors Explaining Community Variations
A cluster analysis of the sediment samples showed similar trends for bacterial and archaeal communities; the shallow sediment layers (1–40cm) of the S8.1 and S8.2 samples showed strong clustering, and the deep sediment layers (50–200cm) of the S8.2 sample also showed strong clustering (Fig.5). The results also revealed that sediment depth was one of the factors contributing to the variability in community structure. Notably, the bacterial community in the surface sediment of S8.2 was similar to that of the 20-cm layer of S8.1, while the archaeal community in the surface sediment of S8.2 was similar to that of the 20-cm and 40-cm sediment layers of S8.1. This similarity is because certain shallow sediment layers in S8.2 were lost when collection was performed using a gravity corer.
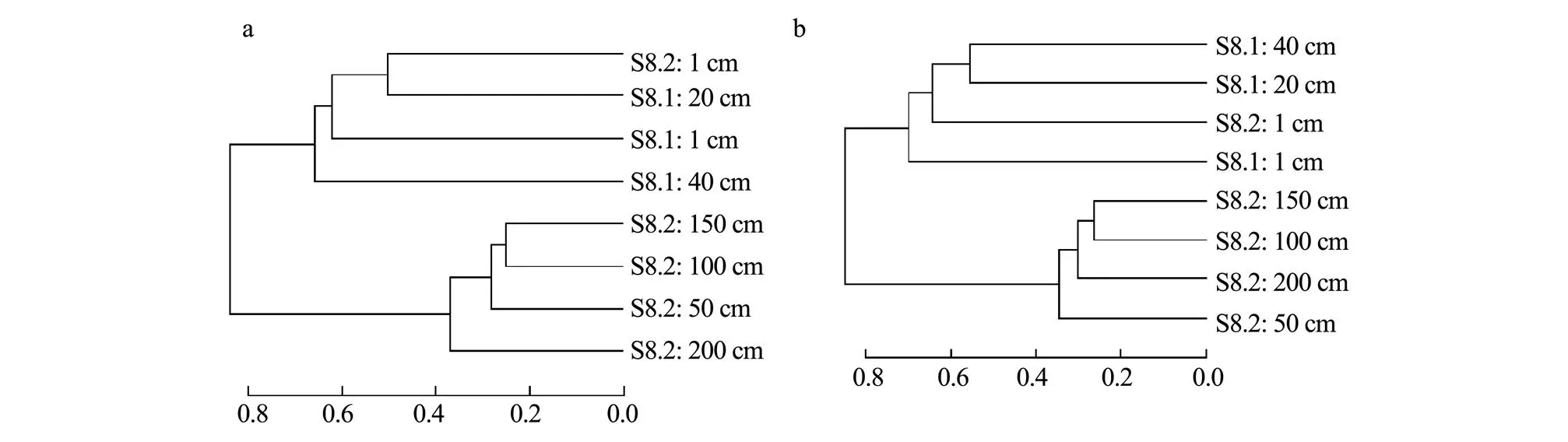
Fig.5 Bray-Curtis clustering in different sediment samples at the OTU level. a, Bacterial communities; b, Archaeal communities.

Fig.6 Redundancy analysis (RDA) biplot of environmental parameters and microbial community structure at the OTU level in different sediment samples. a, Bacterial community; b, Archaeal community.

Fig.7 Spearman analysis of variation in the abundance and diversity of bacterial and archaeal communities with environmental factors.
Correlations between the environmental parameters and the bacterial and archaeal communities were analyzed by redundancy analysis (RDA) (Fig.6). Bacterial and archaeal communities in different sediment samples were structured by distinct environmental factors. For bacteria, TOC (2=0.98,=0.001), sediment depth (2=0.78,=0.022) and pH (2=0.72,=0.03) were the most significant factors in explaining the total variation (Fig.6a). For archaea, TOC (2=0.93,=0.004), sediment depth (2=0.72,=0.044) and NH4+(2=0.68,=0.048) were significantly influential factors (Fig.6b).
Spearman’s analysis showed that TOC, NO2−and NH4+were positively correlated with the abundances of both bacteria and archaea (<0.05), while the pH and sediment depth showed the opposite trend (<0.05) (Fig.7). For the bacterial communities, sediment depth and pH were negatively correlated with alpha diversity indices (<0.05), including Shannon, Simpson and Chao1 indices. However, TOC, NO2−and NH4+were positively correlated with the Shannon index of the bacterial community (<0.05) (Fig.7). However, no environmental factor was found to have a significant correlation with alpha diversity indices of the archaeal community (Fig.7).
3.5 Metabolic Profiles of Microbial Communities Based on Metagenomics Analysis
In this study, more than 20 Gb of sequencing data were generated for the sediment samples. After trimming, there were 10125.84 and 11710.60 Mb of sequencing data that remained for the 1-cm and 40-cm samples, respectively. By assembly and gene prediction, 656194 (808188 ORFs; average length, 377.03bp) and 766739 (944967 ORFs; ave-rage length, 375.23bp) scaffolds were obtained for the 1-cm and 40-cm samples of S8.1, respectively (Table 4).

Table 4 Sequencing, assembly and annotation statistics
3.5.1 Genes involved in nitrogen metabolism
The detected genes were involved in five nitrogen metabolism pathways: nitrate reduction, dissimilatory nitrate reduction, denitrification, nitrogen fixation, and nitrification pathways (Fig.8a). The assimilatory nitrate reduction pathway includes the,,,,andgenesGenes including,andwere detected in the two sediment samples but had lower relative abundance. Theandgene product catalyzes the first step in this pathway, specifically the conversion of nitrate to nitrite, and thegene product catalyzes the nitrite to ammonium reaction (Fig.9). Deltaproteobacteria and Planctomycetes contributed most toin the studied sediments (Fig.9a). Members that contributed to thegene were Gammaproteobacteria, Actinobacteria and Verrucomicrobia in the two samples.of Nitrospinae andof Nitrospirae contributed a large proportion toexpression in both sediment samples (Fig.9b). The dissimilatory nitrate reduction pathway includes the,,, andgenes. All these genes were detected in the two sediment samples but had lower abundance (Fig.8a). Proteobacteria, including Deltaproteobacteria, Gammaproteobacteria and Alphaproteobacteria, contributed mostly to the,,andgenes (Fig.9a). However, Planctomycetes, Verrucomicrobia and Actinobacteria also contributed a large proportion to,,, and(Fig.9a). Biological denitrification is a key process in the marine nitrogen cycle in which NO3−is converted to gaseous products, including NO, N2O and N2. This pathway includes the,,,, andgenes. In the denitrification pathway, all genes were present in the two sediment samples and were higher in abundance (Fig.8a). Proteobacteria, including Deltaproteobacteria, Gammaproteobacteria, Alphaproteobacteria and Betaproteobacteria, were the most important contributors of all genes involved in this pathway, whereas other members, such as Planctomycetes, Chlorobi, and Bacteroidetes, were also shown to play important roles in denitrification (Fig.9a). Importantly, archaea Thaumarchaeota contributed significantly to the presence ofin S8.1 and S8.2 (Fig.9a). The nitrogen fixation pathway was not active in the sediment samples, and only thegene was detected in the two samples (Fig.8a).andwere the most important contributors to this pathway (Fig.9b). The nitrification pathway includes the microbial oxidation of ammonia to nitrite and subsequently to nitrate. Ammonia oxidation comprises two steps: the first step, the oxidation of ammonia to hydroxylamine (NH2OH), is catalyzed by ammonia monooxygenase (Amo), and hydroxylamine oxidoreductase (Hao) then further oxidizes NH2OH to NO (Caranto and Lancaster, 2017). The genes,, andwere mainly expressed by Thaumarchaeota, Crenarchaeota and Betaproteobacteria in two sediment samples, whereaswas mainly expressed bywithin the Planctomycetes phylum (Fig. 9b). Theandgenes encode products that catalyze the conversion of nitrite to nitrate in the nitrification pathway. Chlorobi and Candidatus Acetothermia were the most important contributors ofandrespectively, whereas Gammaproteobacteria also contributed a large proportion toandgenes (Fig.8a). However, the anaerobic ammonium oxidation (anammox) pathway, which seems to be of ecological importance in marine environments (Oshiki., 2016), was not detected in the two sediment samples. These results indi-cated that denitrification and nitrification pathways were active in the microbial communities of our studied sediments and played important roles in the nitrogen cycles. In addition, the relative abundances of most genes in- volved in nitrogen metabolism other than,andwere higher in the 1-cm layer than in the 40-cm layer, implying that the nitrogen metabolism might more active in the surface layer.

Fig.8 Heatmap of log10 fold change in relative abundance of functional genes involved in nitrogen metabolism (a) and sulfur metabolism (b) based on deep sequencing data obtained from the sediment samples.
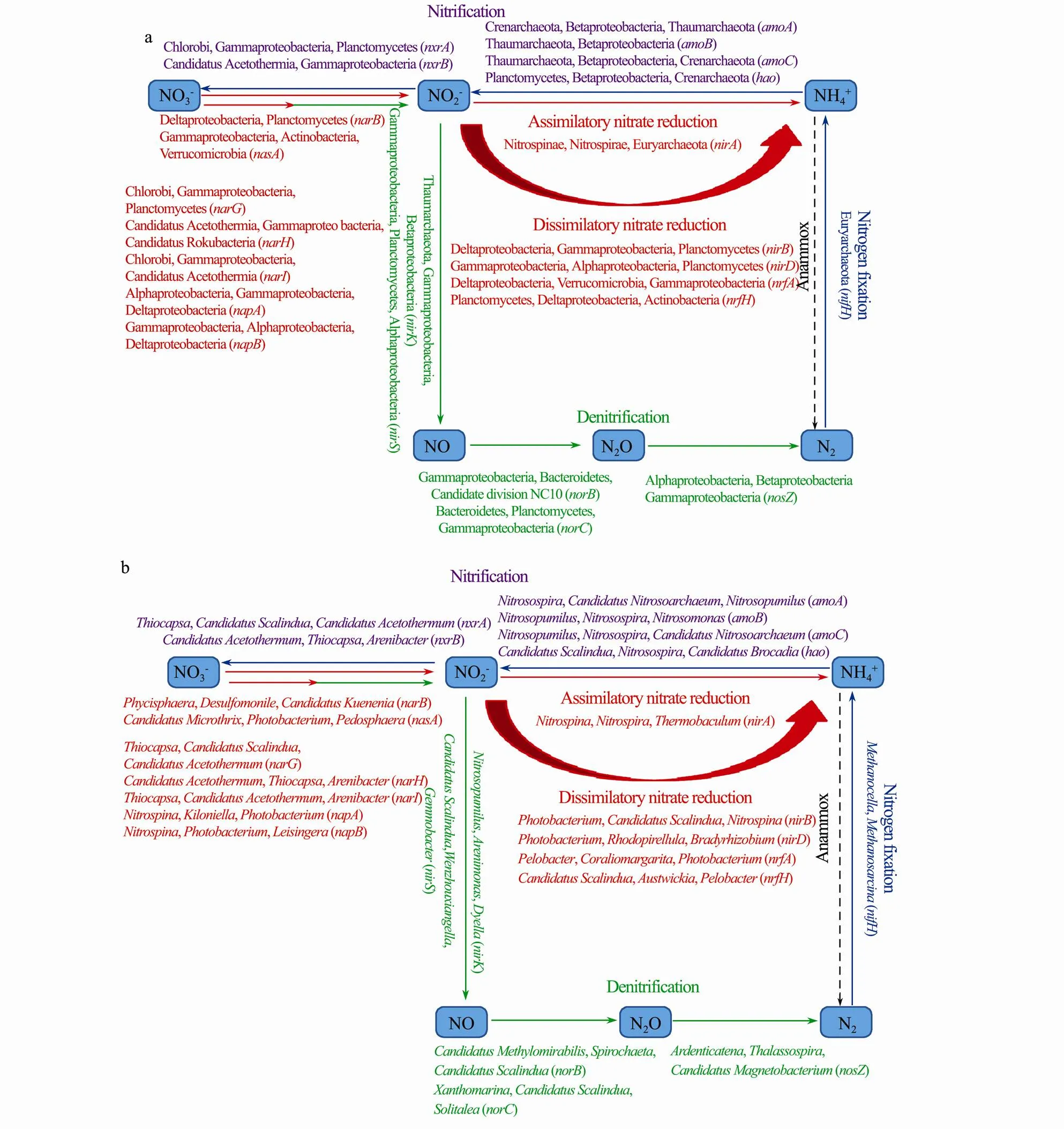
Fig.9 Schematic representation of nitrogen metabolism. The top three abundant phyla (a) and genera (b) that importantly contributed to the different genes are indicated.
3.5.2 Genes involved in sulfur metabolism
According to the KEGG database, the sulfur metabolism includes assimilatory sulfate reduction, dissimilatory sulfate reduction and oxidation and SOX (sulfur oxidation) systems Assimilatory sulfate reduction is a pathway used by a wide range of organisms to convert inorganic sulfate to sulfide, which is further incorporated into the biosynthesis of S-containing amino acids (Brunold, 1993). In this pathway, sulfate is first activated by the catalysis ofandgene products, forming adenosine 5’-pho- sphosulfate (APS). Then, APS is reduced to sulfite by thegene products and further reduced to sulfide byandgene products. All of the above genes were detected in the two sediment samples (Fig.8b). Proteobacteria contributed the most to all these functional genes (Fig.10a). Moreover,within Planc- tomycetes also contributed a large proportion togenes, and Thaumarchaeota contributed a large proportion to,and. (Fig.10). The dissimilatory sulfate reduction and oxidation pathways include,and. Key genes (and) potentially enabling reverse sulfite dissimilatory reduction and potentially involved in the oxidation of sulfide to sulfite were identified in both sediment samples. Deltaproteobacteria and Gammaproteobacteria of Proteobacteria contributed mostly to, while Alphaproteobacteria, Deltaproteobacteria and Planctomycetes contributed mostly to(Fig.10a). The Sox (sulfur oxidation) system is a well-known sulfur oxidation pathway that is catalyzed by the Sox enzyme system. The model of the Sox enzyme system comprises the four periplasmic complexes SoxXA, SoxYZ, SoxB and SoxCD, which catalyze thiosulfate oxidation (Meyer., 2007a). Proteobacteria, including Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria, contributed most to the genes encoding the Sox enzyme system. Moreover, candidate division NC10 contributed significantly to the presence of, whereas Acidithiobacillia and Aquificae also contributed significantly to the presence ofandin the sediment samples (Fig.10a).
4 Discussion
4.1 Bacterial and Archaeal Abundance in the Sediments
Bacteria were more abundant than archaea in the various marine environments (Schippers., 2006; Schippers., 2012; Liu., 2015), as confirmed by the results of the present study. The trends observed in this study for microbial abundance, which decreased with increasing depth, were consistent with the global distribution pattern of benthic microbial abundance (Kallmeyer., 2012). In this study, the bacterial and archaeal 16S rRNA gene abundances were positively correlated with NH4+(< 0.01), NO2−and TOC (<0.05) but negatively correlated with pH (<0.01). Organic matter greatly constrains microbial abundance because this abundance highly depends on organic matter as a carbon source and main electron donor (Zhou., 2002; Drenovsky., 2004).

Fig.10 Sulfur metabolism in the S8.1 samples. The top three abundant phyla (a) and genera (b) that importantly contributed to the different genes are indicated.
4.2 Shift in Microbial Community Structure with Depth and Potential Environmental Drivers
Our study revealed that the microbial community structure shifted significantly with increasing depth in the studied sediment. Bacterial communities were significantly influenced by sediment depth, TOC and pH, while archaeal communities were significantly influenced by sediment depth, TOC and NH4+. For the bacterial community, Deltaproteobacteria and Gammaproteobacteria were the most abundant classes of Proteobacteria, and their relative abundance generally decreased with depth. Groups within Deltaproteobacteria are well known as sulfate-reducing bacteria (SRBs) and can incompletely oxidize products to acetate or completely oxidized products such as carbon dioxide. As the availability of fresh organic matter decreases with sediment depth, SRBs might be comparably less competitive than other metabolic guilds in the anaerobic food chain and thus decrease with depth (Jochum., 2017). However, predominant bacterial groups Chloroflexiwere believed to have an anaerobic and heterotrophic lifestyle that depends on the recalcitrant organic matter buried in deep sediments (Oni., 2015), leading to a higher relative abundance in the deeper sediments. Gammaproteobacteria in this study were mainly comprised of Woeseiaceae and Halomonadaceae. The Woeseiaceae were mostly represented by the type genus, and Halomonadaceae was mostly represented by the type genusin this study.showed a significantly positive relationship with TOC (=0.778,<0.05), which agreed with its chemoheterotrophic lifestyle using organic molecules as energy and carbon sources (Mußmann., 2017).preferred relatively oligotrophic environments (Kesh- tacher-Liebso., 1995), as evidenced by the significantly negative relationship with TOC in our study (= −0.731,<0.05). In addition, pH was regarded as another environmental parameter that determines the bacterial community according to RDA. Krause. (2012) found that moderate changes in pH can cause shifts in bacterial communities. pH may affect the bacterial community through direct physiological mechanisms and may also reflect the indirect influences of other unmeasured factors (Yannarell., 2005; Krause., 2012).
The archaeal community also showed a clear stratified distribution, which was confirmed by RDA. The relative abundances of Nitrososphaeria and Woesearchaeota were enriched in the shallow sediments. The class Nitrososphaeria, which is mainly involved with aerobic oxidization of ammonium, was enriched in the surface layer and soon decreased with increasing depth. Similarly, Wo- esearchaeota (formerly the Deep-sea Hydrothermal Vent Euryarchaeotic Group 6, DHVEG 6) prefer to grow in relatively oxidative environments (Nunoura., 2012) and were indicated to play a role in organic mineralization (Lin., 2014). Thus, oxygen or redox state might have been the crucial factor contributing to the differences between shallow and deep layer communities. Compared to bacteria, archaea have major importance in the prokaryotic community structure in deep layer, where less-reactive organic matter is buried. It has been proposed that archaea can be united by a universal ecological ability to cope with energy stress (Gold, 1992). In the present study, Bathyarchaeia and Thermoplasmata were enriched in the deep layers and increased accordingly with depth. The phylum Bathyarchaeota, formerly named the Miscellaneous Crenarchaeotal Group, is widespread and abundant in marine subseafloor sediments and is composed of important components of anaerobic microbial communities (Kubo., 2012). Studies have shown that these microbes can anaerobically utilize detrital proteins (Lloyd., 2013), aromatic compounds (Meng., 2014), lignin (Yu., 2017) or other buried organic matter as growth substrates for biosynthesis and energy production. Microorganisms from the archaeal class Thermoplasmata were found to be linked to metha- nogenic activities (Iion., 2013; Poulsen., 2013). The increased abundance of these groups suggested that they might play an important role in the anaerobic degradation of organic carbon in the studied sediments.
4.3 Metabolic Profile
4.3.1 Nitrogen metabolism
The ocean’s nitrogen cycle is driven by complex microbial transformations, including nitrogen fixation, assimilation, and loss. However, genes of nitrogen fixation and assimilatory nitrate reduction had lower abundance (Fig.8a), suggesting that these two pathways might not be active in the studied sediment. Gammaproteobacteria, Deltaproteobacteria, Alphaproteobacteria and Betaproteobacteria of the Proteobacteria were found to play the most important role in the nitrogen cycle. These microbial groups contributed most to functional genes involved in nitrification, denitrification, assimilation and dissimilatory nitrate reduction. In this study,within the phylum Planctomycetes was the most abundant bacterial genus revealed by 16S rRNA gene sequencing.is often described as a marine anammox bacterial genus because the 16S rRNAs of anammox bacteria retrieved from marine ecosystems are exclusively affiliated with this bacterial group (Schmid., 2007; Woebken., 2008; Dang., 2013). However, according to the metagenomic analysis in our study, no genes affiliated with the anammox pathway were detected, implying that anammox might not be active in the studied sedimentsIn contrast,metagenomic analysisin our study showed thatcontributed most to the genes,,,,,and, which are involved in nitrification, dissimilatory nitrate reduction and denitrification. This observation is similar to those reported from metagenome analysis for ‘Scalindua’ species, in which functional genes such as,andwere discovered in these microbes (van de Vossenberg., 2013; Oshiki., 2017; Rambo., 2019). In summary, it was confirmed that the most abundant genus,Scalindua, mightplay a more important role in nitrification, dissimilatory nitrate reduction and denitrification in nitrogen metabolism than anammox. In the nitrification pathway, Crenarchaeota contributed a large proportion of thegene, and Thaumarchaeota contributed most to theandgenes in the two sediment samples (Fig.9a), implying that archaea are important players in the nitrification pathway in our study.
4.3.2 Sulfur metabolism
In our study, metagenomic analysis revealed that Proteobacteria contributed most to the functional genes involved in sulfur metabolism, suggesting that these microbes possibly played an important role in sulfur reduction and oxidation. This observation was in line with the previous observations that Proteobacteria dominated the sulfate-reducing prokaryote (SRP) and sulfur-oxidizing prokaryote (SOP) communities analyzed by functionalgene (..,,and) sequencing (Meyer., 2007b; Jiang., 2009; Zhang., 2017). However, Thaumarchaeota andScalindua (Planctomycetes phylum,Brocadiales order) were also found to contribute a large proportion of functional genes involved in assimilatory sulfate reduction (Fig.10b). A previous report found that two Thaumarchaeota genomes recovered from metagenomic data in the Amazon River can encode a complete assimilatory sulfate reduction pathway (Pinto., 2020). Another previous report predicted Candidatus Brocadiales within Planctomycetes to be an important and unrecognized versatile player in C, N and S cycling in anoxic groundwater (Starke., 2017). It is noteworthy that we identified Thaumarchaeota andScalindua as potential sulfate reducers in the deep-sea sediments of the SCS for the first time, which expands our knowledge of the taxonomic distribution of these microbes in deep-sea systems.
5 Conclusions
In summary, the microbial communities and the potential functions of these communities in sediments from the PRMB in the SCS were analyzed. Proteobacteria, Nitrososphaeria and Woesearchaeota were dominant in the shallow sediments, while Chloroflexi and Bathyarchaeota were dominant in the deep sediments (50-200cm). According to metagenomics analysis, we confirmed that Proteobacteria contributed most to almost all pathways in nitrogen and sulfur metabolism. Moreover, Thaumarchaeota andScalindua also play a vital role in some pathways of nitrogen and sulfur metabolism.
Acknowledgements
We thank Mr. Hong Qiu for analyzing the TOC content. We are also grateful to all staff on thefor assistance with the collection of samples and geochemical data during the cruise. This work was supported by the National Natural Science Foundation of China (Nos. 41620104001 and 41806131) and the Scientific and Technological Innovation Project of the Qingdao National Laboratory for Marine Science and Technology (No. 2016 ASKJ02).
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., Mills, D. A., and Caporaso, J. G., 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing., 10 (1): 57-59.
Brochier-Armanet, C., Boussau, B., Gribaldo, S., and Forterre, P., 2008. Mesophilic Crenarchaeota: Proposal for a third archaeal phylum, the Thaumarchaeota., 6 (3): 245-252.
Brunold, C., 1993. Regulatory interactions between sulfate and nitrate assimilation. In:. De Kok, L. J.,., eds., SPB Academic Publishing, the Hague, the Netherlands, 61-75.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., and Costello, E. K., 2010. QIIME allows analysis of high-throughput community sequencing data., 7 (5): 335-336.
Caranto, J. D., and Lancaster, K. M., 2017. Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase.,114 (31): 8217-8222.
Coolen, M. J. L., Cypionka, H., Sass, A. M., Sass, H., and Overmann, J., 2002. Ongoing modification of Mediterranean Pleistocene sapropels mediated by prokaryotes., 296 (5577): 2407-2410.
Coolen, M. J., Hopmans, E. C., Rijpstra, W. I., Muyzer, G., Schouten, S., Volkman, J. K., and Damsté, J. S., 2004. Evolution of the methane cycle in Ace Lake (Antarctica) during the Holocene: Response of methanogens and methanotrophs to environmental change., 35 (10): 1151- 1167.
Dang, H., Zhou, H., Zhang, Z., Yu, Z., Hua, E., Liu, X., and Jiao, N., 2013. Molecular detection ofScalindua pacif- ica and environmental responses of sediment anammox bacterial community in the Bohai Sea, China., 8 (4): e61330.
Drenovsky, R. E., Vo, D., Graham, K. J., and Scow, K. M., 2004. Soil water content and organic carbon availability are major determinants of soil microbial community composition., 48 (3): 424.
Gold, T., 1992. The deep, hot biosphere.,89 (13): 6045-6049.
Gong, J., Sun, X., Lin, Z., Lu, H., and Lu, Y., 2017. Geochemical and microbial characters of sediment from the gas hydrate area in the Taixinan Basin, South China Sea., 36 (9): 52-64.
He, H., Zhen, Y., Mi, T., Xu, B., Wang, G., Zhang, Y., and Yu, Z., 2015. Community composition and distribution of sulfate- and sulfite-reducing prokaryotes in sediments from the Changjiang Estuary and adjacent East China Sea., 165: 75-85.
Hori, T., Kimura, M., Aoyagi, T., Navarro, R. R., Ogata, A., Sakoda, A., Katayama, Y., and Takasaki, M., 2014. Biodegradation potential of organically enriched sediments under sulfate- and iron-reducing conditions as revealed by the 16S rRNA deep sequencing., 12 (4): 357-366.
Hou, L., Zheng, Y., Liu, M., Gong, J., Zhang, X., Yin, G., and You, L., 2013. Anaerobic ammonium oxidation (anammox) bacterial diversity, abundance, and activity in marsh sediments of the Yangtze Estuary., 118 (3): 1237-1246.
Hug, L., Castelle, C., Wrighton, K., Thomas, B., Sharon, I., Frischkorn, K., 2013. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phy- lum and indicate roles in sediment carbon cycling., 1 (1): 22.
Iino, T., Tamaki, H., Tamazawa, S., Ueno, Y., Ohkuma, M., Suzuki, K., Igarashi, Y., and Haruta, S., 2013.Methanogranum caenicola: A novel methanogen from the anaerobic digested sludge and proposal offam. novandord. nov. for a methanogenic lineage of the class., 28: 244-250.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B., 2006. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin., 103 (8): 2815-2820.
Inagaki, F., Suzuki, M., Takai, K., Oida, H., Sakamoto, T., Aoki, K., Nealson, K. H., and Horikoshi, K., 2003. Microbial com- munities associated with geological horizons in coastal subseaflor sediments from the Sea of Okhotsk., 69 (12): 7224-7235.
Jiang, L., Zheng, Y., Peng, X., Zhou, H., Zhang, C., Xiao, X., and Wang, F., 2009. Vertical distribution and diversity of sulfate-reducing prokaryotes in the Pearl River estuarine sediments, Southern China., 70 (2): 249-262.
Jiao, L., Su, X., Wang, Y., Jiang, H., Zhang, Y., and Chen, F., 2015. Microbial diversity in the hydrate-containing and-free surface sediments in the Shenhu area, South China Sea., 6 (4): 627-633.
Jochum, L. M., Chen, X., Lever, M. A., Loy, A., Jørgensen, B. B., Schramm, A., and Kjeldsen, K. U., 2017. Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay., 83 (23): e01547-17.
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C., and D’Hondt, S., 2012. Global distribution of microbial abundance and biomass in subseafloor sediment.,109 (40): 16213-16216.
Keshtacher-Liebso, E., Hadar, Y., and Chen, Y., 1995. Oligo- trophic bacteria enhance algal growth under iron-deficient conditions., 61 (6): 2439-2441.
Könneke, M., Bernhard, A. E., de la Torre, J. R., Walker, C. B., Waterbury, J. B., and Stahl, D. A., 2005. Isolation of an autotrophic ammonia-oxidizing marine archaeon., 437: 543-546.
Krause, E., Wichels, A., Giménez, L., Lunau, M., Schilhabel, M. B., and Gerdts, G., 2012. Small changes in pH have direct effects on marine bacterial community composition: A microcosm approach., 7 (10): e47035.
Kubo, K., Lloyd, K. G., Biddle, J. F., and Amann, R., 2012. Archaea of the miscellaneous crenarchaeotal group are abundant, diverse and widespread in marine sediments., 6 (10): 1949-1965.
Lenk, S., Arnds, J., Zerjatke, K., Musat, N., Amann, R., and Mußmann, M., 2011. Novel groups ofcatalyse sulfur oxidation and carbon fixation in a coastal, intertidal sediment., 13 (3): 758- 774.
Lin, L. H., Wu, L. W., Cheng, T. W., Tu, W. X., Lin, J. R., Yang, T. F., Chen, P. C., Wang, Y., and Wang, P. L., 2014. Distributions and assemblages of microbial communities along a sediment core retrieved from a potential hydrate-bearing region offshore southwestern Taiwan., 92: 276-292.
Liu, J., Liu, X., Wang, M., Qiao, Y., Zheng, Y., and Zhang, X., 2015. Bacterial and archaeal communities in sediments of the north Chinese marginal seas., 70 (1): 105- 117.
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., Stepanauskas, R., Richter, M., Kleindienst, S., Lenk, S., Schramm, A., and Jørgensen, B. B., 2013. Predominant archaea in marine sediments degrade detrital proteins., 496 (7444): 215.
Magoč, T., and Salzberg, S. L., 2011. FLASH: Fast length adjustment of short reads to improve genome assemblies., 27 (21): 2957-2963.
Mahmoudi, N., Robeson, M. S., Castro, H. F. C., Fortney, J. L., Techtmann, S. M., Joyner, D. C., Paradis, C. J., Pfiffner, S. M., and Hazen, T. C., 2015. Microbial community composition and diversity in Caspian Sea sediments., 91 (1): 1-11.
McCaig, A. E., Phillips, C. J., Stephen, J. R., Kowalchuk, G. A., Harvey, S. M., Herbert, R. A., Embley, T. M., and Prosser, J. I., 1999. Nitrogen cycling and community structure of proteobacterial β-subgroup ammonia-oxidizing bacteria within polluted marine fish farm sediments., 65 (1): 213-220.
Meng, J., Xu, J., Qin, D., He, Y., Xiao, X., and Wang, F., 2014. Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses., 8 (3): 650-659.
Meyer, B., and Kuever, J., 2007. Molecular analysis of the diversity of sulfate-reducing and sulfur-oxidizing prokaryotes in the environment, usingas functional marker gene., 73 (23): 7664-7679.
Meyer, B., Johannes, F., and Kuever. J., 2007. Molecular analysis of the distribution and phylogeny of thegene among sulfur-oxidizing bacteria – Evolution of the Sox sulfur oxidation enzyme system., 9 (12): 2957-2977.
Mußmann, M., Pjevac, P., Krüger, K., and Dyksma, S., 2017. Genomic repertoire of the/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments., 11 (5): 1276-1281.
Nunoura, T., Takaki, Y., Kazama, H., Hirai, M., Ashi, J., Imachi, H., and Takai, K., 2012. Microbial diversity in deep-sea methane seep sediments presented by SSU rRNA gene tag sequencing., 27 (4): 382-390.
Oni, O. E., Schmidt, F., Miyatake, T., Kasten, S., Witt, M., Hinrichs, K. U., and Friedrich, M. W., 2015. Microbial communities and organic matter composition in surface and subsurface sediments of the Helgoland mud area, North Sea., 6: 1290.
Oshiki, M., Mizuto, K., Kimura, Z. I., Kindaichi, T., Satoh, H., and Okabe, S., 2017. Genetic diversity of marine anaerobic ammonium-oxidizing bacteria as revealed by genomic and proteomic analyses of ‘Scalindua japonica’., 9 (5): 550-561.
Oshiki, M., Satoh, H., and Okabe, S., 2016. Ecology and physiology of anaerobic ammonium oxidizing bacteria., 18 (9): 2784-2796.
Peiffer, J. A., Spor, A., Koren, O., and Jin, Z., 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions., 110 (16): 6548-6553.
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y., 2010. IDBA–A practical iterative de Bruijn graphassembler. In:. Springer, Berlin, Heidelberg, 426-440.
Pinto, O. H., Silva, T. F., Vizzotto, C. S., Santana, R. H., Lopes, F. A., Silva, B. S., Thompson, F. L., and Kruger, R. H., 2020. Genome-resolved metagenomics analysis provides insights into the ecological role of Thaumarchaeota in the Amazon River and its plume., 20 (1): 13.
Porat, I., Vishnivetskaya, T. A., Mosher, J. J., Brandt, C. C., Yang, Z. K., Brooks, S. C., Liang, L. Y., Drake, M. M., Podar, M., Brown, S. D., and Palumbo, A. V., 2010. Characterization of archaeal community in contaminated and uncontaminated surface stream sediments., 60 (4): 784-795.
Poulsen, M., Schwab, C., Jensen, B. B., Engberg, R. M., Spang, A., Canibe, N., Hojberg, O., Milinovich, G., Fragner, L., Schleper, C., Weckwerth, W., Lund, P., Schramm, A., and Urich, T., 2013. Methylotrophic methanogenicimplicated in reduced methane emissions from bovine rumen., 4: 1428.
Rambo, I. M., Dombrowski, N., Constant, L., Erdner, D., and Baker, B. J., 2019. Metabolic relationships of uncultured bacteria associated with the microalgae., 11: 1pp.
Schippers, A., and Neretin, L. N., 2006. Quantification of microbial communities in near-surface and deeply buried marine sediments on the Peru continental margin using real-time PCR., 8 (7): 1251-1260.
Schippers, A., Kock, D., Höft, C., Köweker, G., and Michael, S., 2012. Quantification of microbial communities in subsurface marine sediments of the Black Sea and off Namibia.,3: 16.
Schmid, M. C., Risgaard-Petersen, N., van de Vossenberg, J., Kuypers, M. M. M., Lavik, G., Petersen, J., Hulth, S., Thamdrup, B., Canfield, D., Dalsgaard, T., Rysgaard, S., Sejr, M. K., Strous, M., Op den Camp, H. J. M., and Jetten, M. S. M., 2007. Anaerobic ammonium-oxidizing bacteria in marine environments: Widespread occurrence but low diversity.,9 (6): 1476-1484.
Starke, R., Müller, M., Gaspar M., Marz, M., Küsel, K., Totsche, K. U., von Bergen, M., and Jehmlich, N., 2017. Candidate Brocadiales dominates C, N and S cycling in anoxic groundwater of a pristine limestone-fracture aquifer., 152: 153-160.
van de Vossenberg, J., Woebken, D., Maalcke, W. J., Wessels, H. J., Dutilh, B. E., and Kartal, B., 2013. The metagenome of the marine anammox bacterium ‘Scalindua profunda’ illustrates the versatility of this globally important nitrogen cycle bacterium., 15 (5): 1275- 1289.
Varon-Lopez, M., Dias, A. C. F., Fasanella, C. C., and Durrer, A., 2014. Sulphur-oxidizing and sulphate-reducing communities in Brazilian mangrove sediments., 16 (3): 845-855.
Wasmund, K., Schreiber, L., Lloyd, K. G., Petersen, D. G., Schramm, A., Stepanauskas, R., Jørgensen, B. B., and Adrian, L., 2014. Genome sequencing of a single cell of the widely distributed marine subsurface Dehalococcoidia, phylum Chlo- roflexi., 8 (2): 383-397.
Woebken, D., Lam, P., Kuypers, M. M. M., Naqvi, S. W. A., Kartal, B., Strous, M., Jetten, M. S. M., Fuchs, B. M., and Amann, R., 2008. A microdiversity study of anammox bacteria reveals a novelScalindua phylotype in marine oxygen minimum zones., 10 (11): 3106-3119.
Xie, S. P., Xie, Q., Wang, D., and Liu, W. T., 2003. Summer upwelling in the South China Sea and its role in regional climate variations., 108 (C8): 3261.
Yannarell, A. C., and Triplett, E. W., 2005. Geographic and environmental sources of variation in lake bacterial community composition., 71 (1): 227-239.
Yu, T., Liang, Q., Niu, M., and Wang, F., 2017. High occurrence of Bathyarchaeota (MCG) in the deep-sea sediments of South China Sea quantified using newly designed PCR primers., 9 (4): 374-382.
Zhang, Y., Su, X., Chen, F., Wang, Y., Jiao, L., Dong, H., Huang, Y., and Jiang, H., 2012. Microbial diversity in cold seep sediments from the northern South China Sea., 3 (3): 301-316.
Zhang, Y., Wang, X., Zhen, Y., Mi, T., He, H., and Yu, Z., 2017. Microbial diversity and community structure of sulfate-re- ducing and sulfur-oxidizing bacteria in sediment cores from the East China Sea., 8: 2133.
Zhang, Y., Zhao, Z., Dai, M., Jiao, N., and Herndl, G. J., 2014. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea., 23 (9): 2260-2274.
Zhou, J., Xia, B., Treves, D. S., Wu, L. Y., Marsh, T. L., O’Neill, R. V., Palumbo, A. V., and Tiedje, J. M., 2002. Spatial and resource factors influencing high microbial diversity in soil., 68 (1): 326- 334.
. E-mail:zhenyu@ouc.edu.cn
May 3, 2019;
February 6, 2020;
March 30, 2020
(Edited by Ji Dechun)
杂志排行

Journal of Ocean University of China的其它文章
- Centurial Evolution of an Offshore Mud Deposition Area in the Changjiang (Yangtze) Estuary and Its Links to Environmental and Anthropogenic Activities
- Characteristics and Origins of Suspended Pyrite in the Mixing Zone of the Yangtze Estuary
- Characterization of Fe(III)-Reducing Enrichment Cultures and Isolation of Enterobacter sp. Nan-1 from the Deep-Sea Sediment, South China Sea
- Evolution of Palaeoenvironment of the South Yellow Sea Since the Last Deglaciation
- Sulfate-Methane Transition Depths and Its Implication for Gas Hydrate
- Comprehensive Investigation and Assessment of Nutrient and Heavy Metal Contamination in the Surface Water of Coastal Bohai Sea in China