大豆分枝数相关分子标记开发及qBN-18位点精细定位
2020-09-25吴海涛苏伯鸿LamlomSobhi邱丽娟
吴海涛 张 勇 苏伯鸿 Lamlom F Sobhi 邱丽娟
大豆分枝数相关分子标记开发及位点精细定位
吴海涛1,3,**张 勇2,**苏伯鸿1,3,**Lamlom F Sobhi3,4邱丽娟3,*
1东北农业大学农学院, 黑龙江哈尔滨 150030;2黑龙江省农业科学院克山分院, 黑龙江齐齐哈尔 161606;3农作物基因资源与遗传改良国家重大科学工程/ 农业部种质资源利用重点实验室 / 中国农业科学院作物科学研究所, 北京 100081;4埃及亚历山大大学农业学院Saba Basha植物生产部, 埃及亚历山大市
分枝数是影响大豆产量的重要因素之一, 与植株结荚率直接相关; 同时也是决定大豆株型的重要组成因子, 并通过调节群体结构、种植密度等进一步影响产量。目前关于大豆分枝数QTL (quantitative trait loci)精细定位与图位克隆的报道极少。因此, 发掘参与调控大豆分枝的基因/QTL对于株型建成的基础研究和高产品种培育的应用研究都具有重要意义。本研究在少分枝品种垦丰19 (KF19)与多分枝品种垦农24 (KN24)组合F2的基础上, 培育出由606个株系组成的F7:8重组自交系(recombinant inbred lines, RIL)群体, 以及由1486个单株KF19-BC3F2和1150个单株KN24-BC2F2组成的2个回交群体。在18号染色体分枝数QTL新位点()的定位区间内筛选出多态性SSR标记11个, 利用RIL群体将的定位区间由1.6 Mb缩小到113 kb。在定位区间内开发了2个InDel标记BR69与BR77。进一步利用回交群体筛选交换单株, 将定位区间缩小到63.7 kb, 包括9个基因。本研究结果为大豆分枝数的基因图位克隆及分子标记辅助育种创造了条件。
大豆; 分枝数; QTL定位; 候选基因
大豆是世界重要的粮食和经济作物, 是人类植物脂肪和蛋白质的重要来源, 在日常膳食结构中占有重要地位。目前大豆和其他高产作物相比, 相对产量偏低, 提高大豆产量潜力是大豆育种的重要任务[1]。分枝数与大豆产量密切相关, 是构成大豆产量的重要因子[2]。分枝数影响植株结实数与群体通风透光状况, 进而影响植株对光能的利用, 还可以通过改善株型、调整大豆群体结构间接影响大豆产量[3-4]。同时, 大豆分枝发育对出苗不齐起到补偿作用, 分枝上种子产量的提高, 可以有效弥补单位土地面积主茎种子产量下降引起的损失, 从而补充了群体的产量[5-6]。因此发掘调控大豆分枝发育的相关基因对于培育高产大豆品种具有重要意义。
大豆分枝数由多基因调控, 分枝的发生受遗传因子、激素和环境等多种因素的影响[7]。迄今为止, 利用大豆的F2群体与重组自交系群体定位了数十个大豆分枝数相关QTL[8-21]。其中在SoyBase (https:// www.soybase.org)中已列出21个分枝数QTL[9,18-19,21]。这些QTL大多分布在10个染色体上, 包括4号、5号、6号、10号、11号、14号、15号、17号、18号和19号染色体。其中在不同世代中表现稳定的QTL有11个[13-14,16-17]。在不同环境中检测到2个稳定QTL[15]。虽然定位的大豆分枝数QTL比较多, 但分枝相关基因研究的报道较少, 谭冰等[22]通过Blastp分析183个植物分枝相关基因的氨基酸序列, 在大豆基因组中获得406个同源基因, 经过共定位分析有57个基因位于20个QTL区间内, 其中2个基因位于多次被检测到的QTL区间, 即COBL1的2个同源基因(和)定位在标记Satt294~Satt399之间。Sangrea等[21]对400份大豆品种进行全基因组关联分析(genome-wide association study, GWAS)分析, 并通过少分枝品种Jiyu 69和多分枝品种SS0404-T5-76杂交构建近等基因系群体进行精细定位, 将分枝数QTL ()精细定位到6号染色体的460 kb区间内, 包含13个候选基因, 结合表达分析和基因序列分析推测,()可能是调控大豆分枝发育的候选基因。
本研究以前期定位在18号染色体的分枝数QTL新位点为基础, 以构建的RIL群体和回交群体为材料, 通过鉴定F7:8群体基因型将的定位区间由1.6 Mb缩小到113 kb, 并利用回交群体筛选交换单株将其精细定位在63.7 kb区间内。为大豆分枝数基因克隆、调控机制解析和分子标记选择奠定了基础。
1 材料与方法
1.1 定位群体构建及表型调查
在张霞等[20]以少分枝大豆品种垦丰19 (KF19)和多分枝大豆品种垦农24 (KN24)构建的F2群体基础上, 本研究通过单粒传培育出由606个家系组成的F7:8重组自交系。以KN24为轮回亲本, 与KN24和KF19的F1植株回交2次, 获得17个BC2F1单株, 自交获得BC2F2群体, 共1150个单株。同时, 以KF19为轮回亲本回交3次获得42个BC3F1单株, 自交获得BC3F2群体, 共1486个单株。在黑龙江省农业科学院克山分院试验站种植2个亲本、RIL群体和回交群体。行长3 m, 行距0.4 m, 株距0.1 m。田间按常规管理, 大豆植株成熟后, 去除边缘单株, 参照《大豆种质资源描述规范和数据标准》[23]考种。分枝数为每个植株主茎上的有效分枝数, 分枝上有2个或多个节, 收获时至少有1个成熟的种子荚。2018年与2019年亲本分别考种25株与35株。F7群体每行考种4个单株, F7:8群体每行考种8个单株。采集F7单株叶片, 干冰冷冻后保存在-80℃冰箱备用。
1.2 大豆基因组DNA提取
利用改良的CTAB法提取大豆基因组DNA。取大约100 mg的大豆叶片或组织, 加入2.0 mL离心管中, 到每孔装有一颗5 mm钢珠, 在液氮中迅速冷冻, 放在高通量组织研磨机中打样30 s; 取出后每管加入600 µL CTAB溶液和5 µL RNase A (10 mg mL-1)并振荡混匀5 min; 65℃水浴锅中水浴1 h, 每隔10 min颠倒混匀1次; 水浴完成后加入600 µL酚/氯仿, 轻轻混匀10 min再室温静置10 min, 12,000´离心10 min, 吸取上清; 加入等体积氯仿, 轻轻混匀10 min再室温静置10 min, 12,000´离心10 min, 吸取上清; 加入等体积异丙醇,-20℃静置1 h或过夜; 再12,000´离心10 min, 弃上清; 用75%乙醇洗涤2次, 12,000´离心后弃上清并进行甩干, 用移液器吸除管底残留的乙醇; 风干20 min后加入100 µL灭菌的蒸馏水溶解DNA, 使用NanoDrop 2000/ 2000c超微量分光光度计和0.8%琼脂糖凝胶电泳检测样品DNA浓度和质量。
1.3 分子标记的选择及分析
从SoyBase数据库(https://soybase.org/)中选择位于大豆18号染色体定位区间内的55对SSR标记, 由博迈德生物技术有限公司合成, 用KN24和KF19进行多态性引物筛选(表1)。PCR总反应体系为20 µL, 包括10×Easybuffer 2.0 µL、2.5 mmol L-1dNTPs 1.5 µL、无菌水12.3 µL、Easy酶0.2 µL、引物(2 µmol L-1) 2.0 µL和DNA (70 ng µL-1) 2.0 µL。反应程序为95℃ 5 min; 95℃ 30 s, 55℃ 30 s, 72℃ 30 s, 34个循环; 72℃ 5 min, 4℃保存。用6%非变性聚丙烯酰胺凝胶电泳检测SSR标记的扩增产物。

表1 精细定位所用分子标记
1.4 InDel标记开发与鉴定
根据亲本定位区间随机测序发现, 55,882,277与55,946,270位点分别存在1个InDel标记, 从Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html)下载含InDel变异位点的序列, 利用Primer3web (http://primer3.ut.ee/)网站分别设计包含InDel的标记BR69与BR77的引物(表2)。为检测2个标记的灵敏度, 用特异引物扩增12个不同浓度的KN24与KF19, DNA浓度分别为100、80、60、40、20、10、5、2、1、0.5、0.2、0.1 ng µL-1。PCR总反应体系为20 µL, 包括10×Easybuffer 2.0 µL、2.5 mmol L-1dNTPs 1.5 µL、无菌水12.3 µL、Easy酶0.2 µL、引物(2 µmol L-1) 2.0 µL和DNA 2.0 µL。反应程序为95℃ 5 min; 95℃ 30 s, 54℃ 30 s, 72℃ 30 s, 36个循环; 72℃ 5 min, 4℃保存。用6%非变性聚丙烯酰胺凝胶电泳检测InDel标记的扩增产物。筛选最适DNA浓度用于鉴定RIL群体基因型。
1.5 数据统计分析
利用Microsoft Excel 2016计算KF19与KN24分枝数的平均值与标准差, 利用SAS9.4软件进行单因素方差分析, 利用ggplot2 (R语言程序包)中的vioplot程序分析定位区间内InDel标记BR69与BR77各个基因型对应的分枝数分布。
1.6 分枝数QTL定位及验证
利用软件QTL IciMapping V4.1[24]完备区间作图(inclusive composite interval mapping, ICIM)法对分枝数进行QTL定位, 经1000次排列确定LOD阈值, 当实际求得的LOD值大于LOD阈值时, 就认为该区段存在1个QTL。
1.7 候选基因预测
根据定位区间物理位置查询SoyBase (https:// www.soybase.org/)获得基因注释。使用Phytozome v12.1获得的定位区间内基因的表达模式, 使用Sangerbox生成热图。

表2 两个InDel标记的位置、引物序列及预期扩增片段长度
2 结果与分析
2.1 KN24和KF19及其群体分枝数表型分析
KF19为少分枝材料, KN24为多分枝材料(图1-A)。2018年和2019年KF19平均分枝数分别为0.5和0.7, KN24平均分枝数分别为5.5和6.8 (表3)。KF19和KN24的分枝数性状存在极显著差异, 并且在不同年份间表型稳定, 表明KF19和KN24分别为稳定的多分枝和少分枝品种, 适合作为大豆分枝数QTL定位群体的亲本。KF19×KN24的群体F7和F7:8均有606个株系, 分枝数的变异范围为0~10个, 多分枝特性存在超亲现象。F7、F7:8与回交群体F2(KN24-BC2F2、KF19-BC3F2)的分枝数均呈连续性变异, 符合正态分布(图1)。表明分枝数性状是典型的多基因控制的数量性状。

表3 2018年、2019年KF19和KN24及其F7、F7:8群体的分枝数(BN)统计分析

(图1)
A: 亲本植株; B~C: 分别为2018年与2019年KN24与KF19分枝数表型; D~E: 分别为F7与F7:8分枝数频率分布直方图; F: 2018年回交群体F2分枝数频率分布直方图。垂直虚线表示两亲本表型的平均值和标准差。曲线代表密度图。***:<0.001。
A: parent plant; B–C: KN24 and KF19 branch number phenotypes in 2018 and 2019, respectively; D–E: histogram of frequency distribution of F7and F7:8branch numbers, respectively; F: histogram of the frequency distribution of branch number F2in the backcross population in 2018. Mean value and standard deviation of parental phenotypes are indicated by vertical dotted lines. Curve represents density plot. ***:< 0.001.
2.2 用RIL群体精细定位分枝数相关QTL
张霞等[20]利用垦丰19和垦农24杂交构建的F2群体定位到2个分枝数相关QTL, 分别位于6号和18号染色体上, 其中18号染色体QTL位点()介于标记BARCSOYSSR_18_1777和BARCSOYSSR_18_ 1858之间, 约1.6 Mb, 解释8.0%的遗传变异, 是一个新的QTL位点。为了精细定位, 将垦丰19和垦农24的F2构建成1个由606个株系组成的F7:8重组自交系群体。在标记BARCSOYSSR_18_1777和BARCSOYSSR_18_1858之间选择55个SSR标记分析2个亲本发现, 多态性的SSR标记共有14个。根据物理位置选择均匀分布的11个SSR标记鉴定群体基因型, 利用QTL IciMapping 4.1软件绘制遗传连锁图谱并定位QTL (临界阈值LOD=2.5)(图2), 获得了一个LOD值为19.33的分枝数QTL (表4), 将的定位区间由1.6 Mb缩小至113 kb位于标记BARCSOYSSR_18_1834与BARCSOYSSR_18_1841之间, 解释14.24%的遗传变异, 区间内包含14个基因。
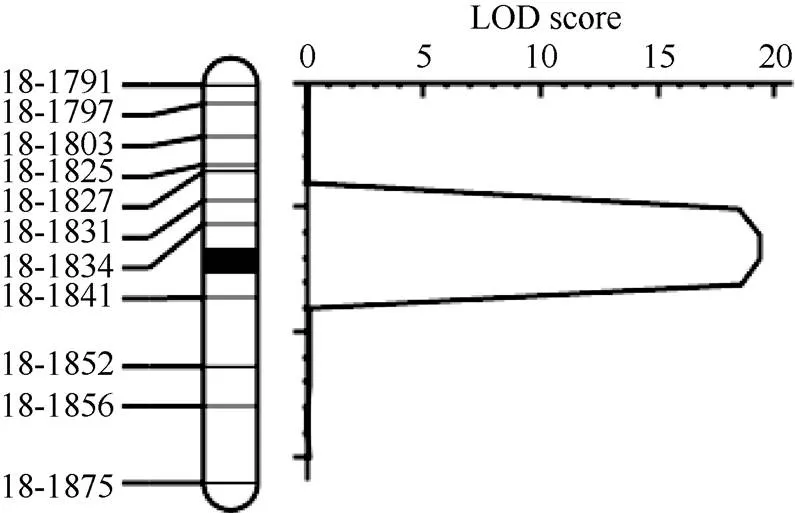
图2 用垦丰19和垦农24构成的RIL群体在大豆18号染色体定位分枝数QTLqBN-18

表4 KF19×KN24的F7:8群体中鉴定到的分枝数QTL
2.3 InDel标记开发与鉴定
2.3.1 InDel标记开发与灵敏度检测 在定位区间内基因间隔区随机测定KF19与KN24 2个亲本序列, 在55,882,277与55,946,270位点分别发现1个InDel位点, 在两亲本间分别有6个、12个碱基的差异, 从Phytozome (https://phytozome.jgi.doe.gov/pz/portal. html)下载含InDel变异位点的序列, 利用Primer3web网站设计BR69与BR77的特异扩增引物(表2)。BR69位于基因的上游, 其物理位置与标记BARCSOYSSR_18_1834相距4.5 kb, 与标记BARCSOYSSR_18_1841相距108.5 kb。BR77位于基因的下游, 其物理位置与标记BARCSOYSSR_18_1834相距68.3 kb, 与标记BARCSOYSSR_18_1841相距44.7 kb。BR69与BR77相距63.7 kb。
为了鉴定所开发的InDel标记的实用性, 设置了12个DNA浓度检测BR69与BR77的灵敏度。BR69在DNA≥2 ng的8个浓度时均可以扩增出两亲本目标条带, 且两亲本间差异明显。在DNA≤1 ng的4个浓度时两亲本均出现杂带, 特异性差。BR77在DNA≥5 ng的7个浓度时均可以扩增出两亲本目标条带, 且两亲本间差异明显, 在DNA≤2 ng的5个浓度时条带不清晰。表明BR69和BR77 2个标记在亲本DNA浓度为5~100 ng µL-1之间均扩增出清晰条带, 可用于群体基因型鉴定。
2.3.2 RIL群体基因型鉴定 用2个InDel标记BR69与BR77鉴定RIL群体基因型。在BR69位点, 多分枝基因型(aa)有290个株系, 平均分枝数为5.22; 少分枝基因型(bb)有274个株系, 平均分枝数为3.80。在BR77位点, 多分枝基因型(aa)有271个株系, 平均分枝数为5.22; 少分枝基因型(bb)有259个株系, 平均分枝数为3.85 (图3)。表明2个标记与分枝数紧密连锁, 可用于分枝数改良的标记辅助育种。

图3 RIL群体中标记BR69与BR77的基因型与表型相关性分析
A: BR69的基因型与表型相关性分析; B: BR77的基因型与表型相关性分析。图中横坐标为基因型, 多分枝记为a, 少分枝记为b, 纵坐标为分枝数。***< 0.001。
A: correlation analysis between genotype and phenotype of BR69; B: correlation analysis between genotype and phenotype of BR77. The abscissa is the genotype, the multiple branches are denoted as a, the few branches are denoted as b, and the vertical ordinate is the number of branches. ***< 0.001.
2.4 利用回交群体验证定位结果
为了进一步验证和缩小定位区间, 分别以两亲本为轮回亲本构建了2个回交群体KN24-BC2F2与KF19-BC3F2, 用两侧标记BARCSOYSSR_ 18_1777与BARCSOYSSR_18_1875鉴定1150株KN24-BC2F2与1486株KF19-BC3F2的基因型, 筛选出在2个标记间基因型不同的单株有568株。然后用4个SSR标记BARCSOYSSR_18_1791、BARCSOYSSR_ 18_1803、BARCSOYSSR_18_1827与BARCSOYSSR_ 18_1856鉴定2个标记之间的重组单株, 其中, 在BARCSOYSSR_18_1827与BARCSOYSSR_18_1856之间发生交换的单株有225个。然后用7个SSR标记BARCSOYSSR_ 18_1825、BARCSOYSSR_18_1831、BARCSOYSSR_ 18_1832、BARCSOYSSR_18_1834、BARCSOYSSR_ 18_1841、BARCSOYSSR_18_1847、BARCSOYSSR_ 18_1852与2个InDel标记BR69、BR77鉴定筛选出来的225个重组单株, 结合表型分析, 最终将由BARCSOYSSR_18_1777与BARCSOYSSR_ 18_1875之间的1.6 Mb缩小到BR69与BR77之间, 约为63.7 kb (图4)。
2.5 候选基因预测
根据定位区间物理位置查询SoyBase (https:// www.soybase.org/)发现, 定位区间内共有9个假定基因, 其中6个基因在SoyBase有基因注释(表5)。从Phytozome v12.1获得这9个基因的表达模式, 用Sangerbox生成热图(图5),在豆荚、种子与花中有表达。与在豆荚、根、种子、茎、叶、茎顶端分生组织(shoot apical meristem, SAM)、花、节、根毛9个组织中均有表达。在根、节、根毛中有表达。在根中有表达。与在各组织中均无表达。在豆荚、根、茎、花、节、根毛中有表达。在根、茎、茎顶端分生组织、花、节、根毛中有表达。
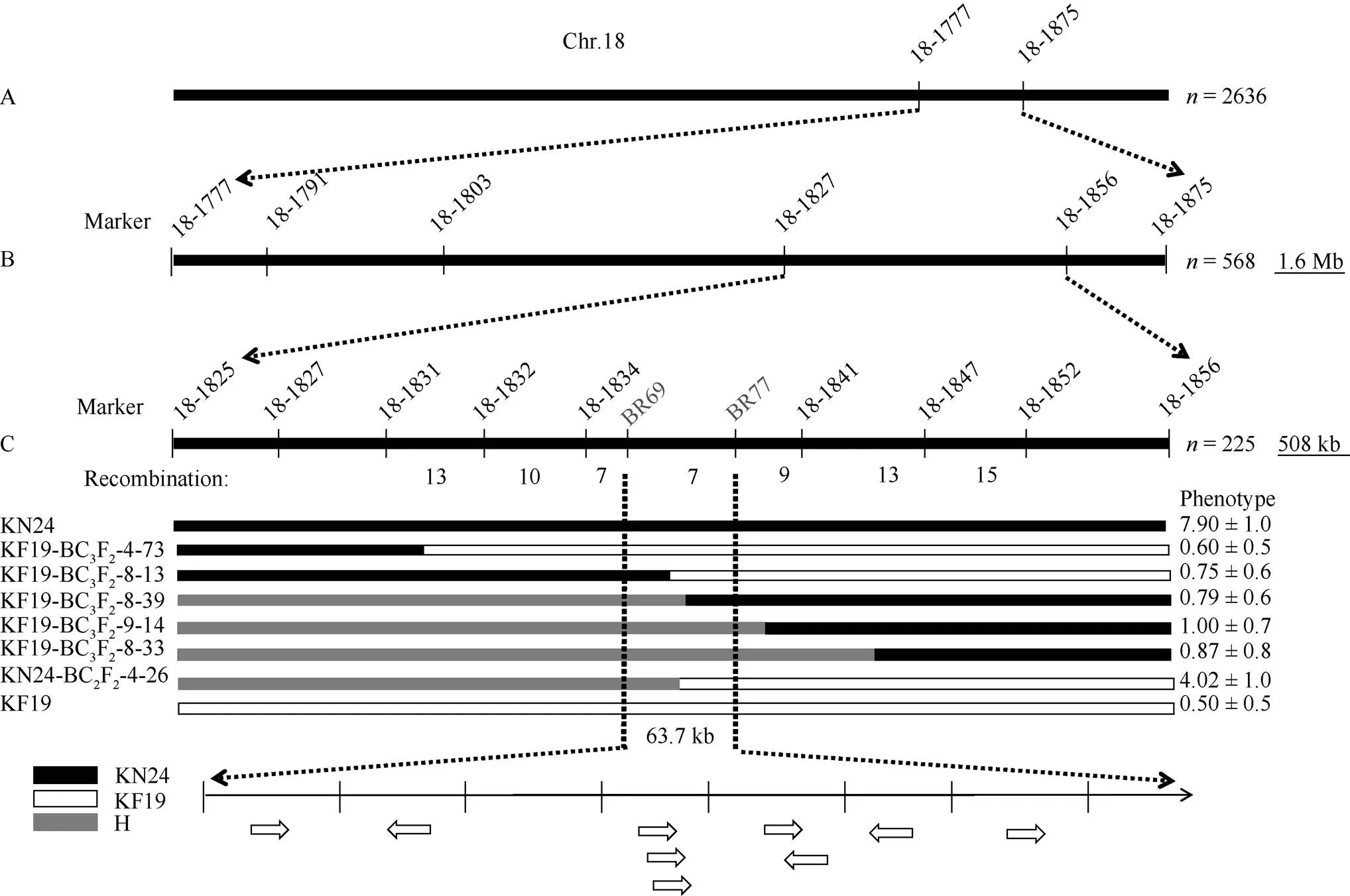
图4 qBN-18位点的验证
A:利用KF19-BC3F2的1486个单株和KN24-BC2F2的1150个单株鉴定了标记BARCSOYSSR_18_1777和BARCSOYSSR_18_1875之间的位点; B:利用BARCSOYSSR_18_1791、BARCSOYSSR_18_1803、BARCSOYSSR_18_1827与BARCSOYSSR_18_1856 4个标记鉴定BARCSOYSSR_18_1777和BARCSOYSSR_18_1875之间的568个重组单株; C:位于标记BR69与标记BR77之间。H: 杂合基因型。
A:loci between marker BARCSOYSSR_18_1777 and BARCSOYSSR_18_1875 was identified using 1486 individuals of KF19-BC3F2and 1150 individuals of KN24-BC2F2; B: BARCSOYSSR_18_1791, BARCSOYSSR_18_1803, BARCSOYSSR_18_1827 and BARCSOYSSR_18_1856 were used to identify 568 recombinant strains between BARCSOYSSR_18_1777 and BARCSOYSSR_18_1875; C:is located between tag BR69 and tag BR77. H: heterozygous genotype.
植物的分枝是由茎顶端分生组织衍生出的腋生分生组织分化来的, 因此初步确定在茎顶端分生组织有表达的3个基因、、可能与分枝数相关。

表5 定位区间内基因的同源基因及基因注释
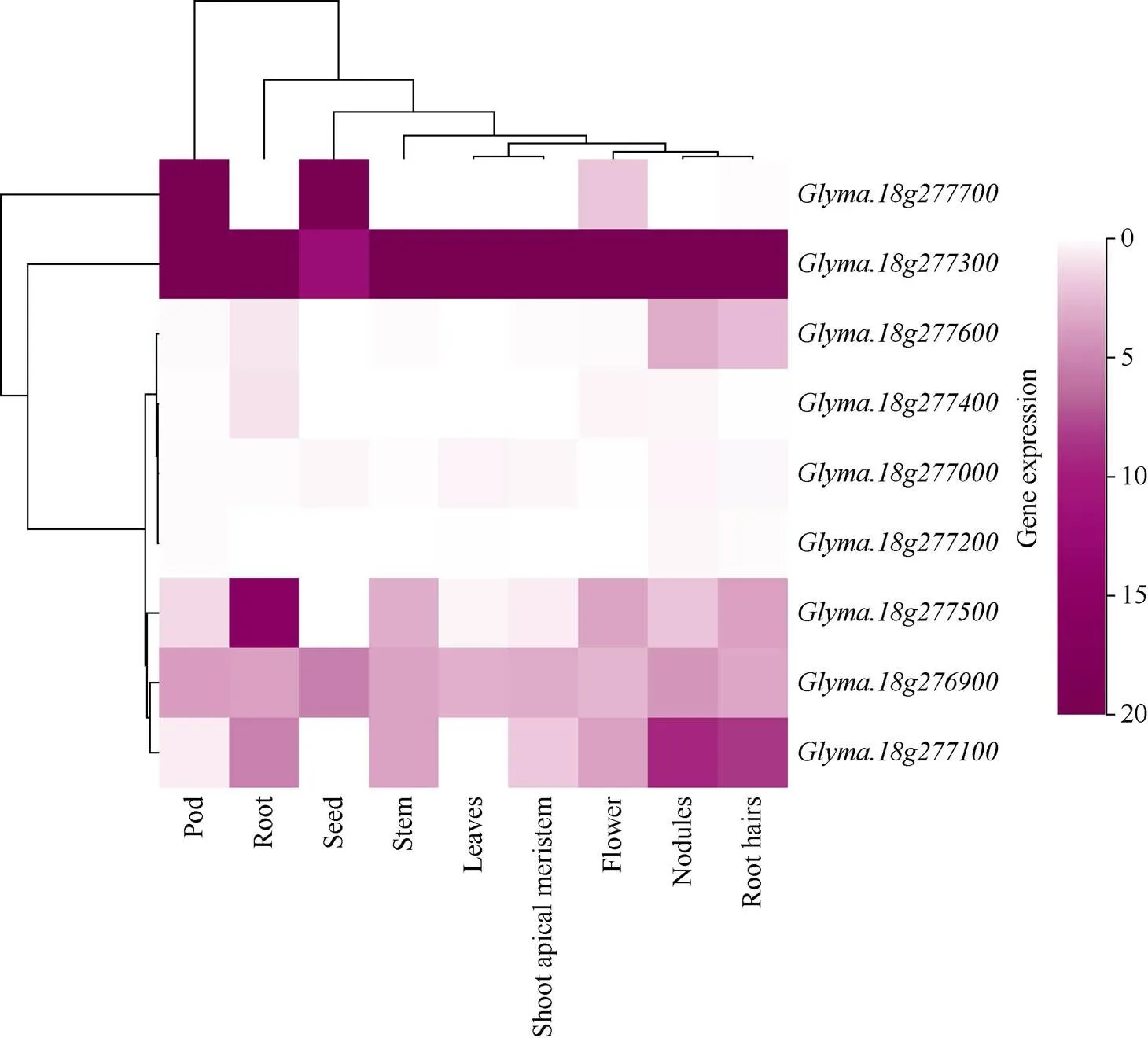
图5 用Phytozome v12.1获得9个基因的表达谱
3 讨论
3.1 RIL群体与回交群体共同定位的优势
QTL定位所用的群体有F2、重组自交系(recombinant inbred lines, RILs)、回交重组自交系(backcross inbred lines, BILs)或双单倍体系(double inbred lines, DHLs), 以及基于多个分子标记生成的连锁图谱[25]。由于QTL分析是建立在统计计算的基础上的, 即使使用大量的群体和许多遗传标记, 也很难确定单个QTL的精确位置, 因此, 精细定位需要其他群体[26]。F2群体为暂时性分离群体, 与RIL群体相比, 其株系内基因型还未达到纯合, 因此不能永久使用[27]。回交导入系是通过与一个亲本多代回交, 并结合系统的标记辅助选择(molecular marker-assisted selection, MAS)产生的。在独特的基因组背景下包含少量供体片段(或QTL)的导入, 有助于对目标QTL进行全面分析, 可以评估QTL在轮回亲本基因组背景下的作用[26]。而且回交群体家系间的遗传背景相似, 供体背景对目标性状QTL效应检测的影响较小, QTL检测更为准确[28]。目前大豆分枝数QTL定位所使用的群体多为F2与RIL群体[8-21]。本研究在利用F2群体进行初定位的基础上, 通过鉴定RIL群体基因型进行精细定位, 用2个回交群体验证定位结果, 经过多重验证, 定位结果可信度更高。
3.2 大豆分枝数QTL精细定位
大豆分枝数是由多基因控制的数量性状[29], 不仅与产量关系密切, 同时也是影响大豆株型的重要因子, 定位并克隆分枝发育相关基因对培育高产大豆新品种具有重要意义。然而, 大豆分枝数的QTL定位研究尚处于起步阶段[22]。虽然已经定位的大豆分枝数QTL较多[8-21]。但精细定位仅有1例, Sangrea等[21]利用近等基因系将6号染色体上的分枝数位点精细定位到的460 kb区间内。
本研究是在张霞等[20]定位分枝数的新QTL基础上, 用11个SSR标记鉴定KN24×KF19 F7:8, 将定位区间缩小到113 kb, 解释14.24%的遗传变异; 并用回交群体将定位区间进一步缩小至63.7 kb, 包含9个基因。该位点尚未见相关报道。因此, 本研究精细定位在18号染色体的大豆分枝数QTL是迄今为止定位区间小、包含候选基因少的QTL, 为该位点的图位克隆提供了重要信息, 也为分子标记选择分枝数创造了条件。
3.3 大豆分枝数候选基因
植物的分枝是由茎顶端分生组织衍生出的腋生分生组织分化来的, 腋芽形成后可能发育形成分枝, 也可能一直保持休眠状态直到生长被激活[30]。腋芽的生长受基因、激素、发育状况和环境等多种因素的影响, 各因素的相互作用共同决定腋芽处于休眠状态还是继续发育[31]。植物的顶端优势会抑制侧芽的生长。由于生长素是在茎顶端合成的, 它被植物的下半部分所吸收, 抑制了分枝的生长, 所以去掉茎顶端后可以消除顶端优势, 促进一个或多个腋芽的生长[32]。考虑到茎顶端分生组织与分枝的发育有关, 用Phytozome v12.1获得的定位区间内9个基因的表达模式, 使用Sangerbox生成热图。其中有3个基因、、在茎顶端分生组织中有表达, 这些基因在本研究群体中的表达情况还有待检测。
根据SoyBase (https://www.soybase.org/)提供的信息对区间内基因进行注释(表5), 其中是一个WPP蛋白家族基因, WPP蛋白家族表达的减少导致拟南芥根系有丝分裂活性降低, 导致根长缩短, 侧根数量减少[33]。是一个bZIP (basic leucine zipper)家族基因。bZIP TFs是一个高度保守家族, 调节植物生长发育过程中的多种现象, 并参与胁迫反应和激素信号传导[34]。许多真核生物(包括酵母、动物和植物)的基因组已经鉴定或预测到了bZIP TFs家族成员[35-37]。在正常条件下, bZIP TFs参与各种生物过程。例如, 它们在器官和组织分化[38]、细胞伸长[39]和体细胞胚发生[40]中起重要作用。它们还在响应各种生物/非生物胁迫和信号(如激素和糖信号)方面发挥重要的调节作用, 如光反应[41]和渗透胁迫[42]。是一个6-磷酸葡萄糖酸脱氢酶(6PGDH)蛋白家族基因。磷酸戊糖途径(PPP)对于植物代谢至关重要[43], 在PPP的氧化部分中, 葡萄糖-6-P脱氢酶(G6PDH)和6-磷酸葡萄糖酸脱氢酶(6PGDH)催化葡萄糖-6-P的不可逆氧化, 产生两分子NADPH和一分子CO2。Gertraud等[44]在玉米叶绿体中定位到了氧化戊糖磷酸途径中6-磷酸葡萄糖酸脱氢酶(6PGDH)功能缺失的2个等位基因, 这些突变导致胚乳种子表型粗糙, 胚乳油和胚乳淀粉减少。目前定位区间内的基因尚无与分枝数相关的报道, 因此候选基因的选择还有待于进一步的鉴定和验证。
4 结论
利用11个SSR标记鉴定到1个由606个株系组成的KF19×KN24F7:8RIL群体基因型并构建遗传图谱, 在18号染色体精细定位到1个大豆分枝数相关QTL, 位于标记BARCSOYSSR_18_1834与BARCSOYSSR_18_1841之间, 解释14.24%的遗传变异。通过开发BR69和BR77 2个InDel标记, 结合回交群体, 将区间从113 kb缩小至63.7 kb。本研究结果为大豆分枝数的基因克隆及分子标记辅助育种创造了条件。
[1] 黄中文, 赵团结, 喻德跃, 陈受宜, 盖钧镒. 大豆产量有关性状QTL的检测. 中国农业科学, 2009, 42: 4155–4165. Huang Z W, Zhao T J, Yu D Y, Chen S Y, Gai J Y. Detection of QTLs of yield related traits in soybean., 2009, 42: 4155–4165 (in Chinese with English abstract).
[2] 刘金刚, 孙恩玉, 曹永强, 刘艳辉, 刘凤丽. 大豆主要生育性状与产量间的关系分析. 杂粮作物, 2005, 25(2): 81–83. Liu J G, Sun E Y, Cao Y Q, Liu Y H, Liu F L. Correlation analysis of the major breeding traits and yield of soybean., 2005, 25(2): 81–83 (in Chinese with English abstract).
[3] 胡珀, 韩天富. 植物茎秆性状形成与发育的分子基础. 植物学通报, 2008, 25(1): 1–13. Hu B, Han T F. Molecular basis of stem trait formation and development in plants., 2008, 25(1): 1–13 (in Chinese with English abstract).
[4] 张永强, 张娜, 王娜, 唐江华, 徐文修, 李亚杰. 种植密度对夏大豆光合特性及产量构成的影响. 核农学报, 2015, 29: 1386–1391. Zhang Y Q, Zhang N, Wang N, Tang J H, Xu W X, Li Y J. Effects of plant population on photosynthetic characteristics and yield components of summer soybean., 2015, 29: 1386–1391 (in Chinese with English abstract).
[5] 董钻, 孙卓韬. 大豆株型、群体结构与产量关系的研究, 第一报: 大豆群体的自动调节和群体内光强、CO2的分布. 大豆科学, 1984, 3(2): 110–119. Dong Z, Sun Z T. Research on the relation of soybean plant form, group structure and output I. The automatic regulation of soybean group and the distribution of light intensity and CO2., 1984, 3(2): 110–119 (in Chinese).
[6] Agudamu, Yoshihira T, Shiraiwa T.Branch development responses to planting density and yield stability in soybean cultivars., 2016, 19: 331–339.
[7] Beveridge C A. Axillary bud outgrowth: sending a message., 2006, 9: 35–40.
[8] 关荣霞. 大豆重要农艺性状的QTL定位及中国大豆与日本大豆的遗传多样性分析. 中国农业科学院博士后研究工作报告, 北京, 2004. Guan R X. QTL Mapping of Soybean Agronomic Characters and Genetic Diversity Analysis of Soybean Cultivars from China and Japan. Post Doctoral Working Repor of Chinese Academy of Agricultural Sciences, Beijing, China, 2004 (in Chinese with English abstract).
[9] 陈庆山, 张忠臣, 刘春燕, 辛大伟, 单大鹏, 邱红梅, 单彩云. 大豆主要农艺性状的QTL分析. 中国农业科学, 2007, 40: 41–47. Chen Q S, Zhang Z C, Liu C Y, Xin D W, Shan D P, Qiu H M, Shan C Y. QTL analysis of major agronomic traits in soybean., 2007, 40: 41–47 (in Chinese with English abstract).
[10] 王珍. 大豆SSR遗传图谱构建及重要农艺性状QTL分析. 广西大学硕士学位论文, 广西南宁, 2004. Wang Z. Construction of Soybean SSR Based Map and QTL Analysis of Important Agronomic Traits. MS Thesis of Guangxi University, Nanning, Guangxi, China, 2004 (in Chinese with English abstract).
[11] 位艳丽. 大豆农艺和品质性状遗传模型分析与QTL定位. 河南农业大学硕士学位论文, 河南郑州, 2011. Wei Y L. Genetic Model Analysis and QTL Mapping of Agronomic and Quality Traits in Soybean. MS Thesis of Henan Agricultural University, Zhengzhou, Henan, China, 2011 (in Chinese with English abstract).
[12] 梁慧珍, 余永亮, 杨红旗, 张海洋, 董薇, 李彩云. 大豆产量及主要农艺性状QTL的上位性互作和环境互作分析. 作物学报, 2014, 40: 37–44. Liang H Z, Yu Y L, Yang H Q, Zhang H Y, Dong W, Li C Y. Epistatic effects and QTL × environment interaction effects of QTLs for yield and agronomic traits in soybean., 2014, 40: 37–44 (in Chinese with English abstract).
[13] 何冉, 关荣霞, 刘章雄, 朱晓丽, 常汝镇, 邱丽娟. 用分离群体中的残余杂合系定位大豆C1连锁群的分枝数位点. 中国农业科学, 2009, 42: 1152–1157.He R, Guan R X, Liu Z X, Zhu X L, Chang R Z, Qiu L J. Mapping thelocus to LG C1 for soybean branching using residual heterozygous lines derived from a segregation population., 2009, 42: 1152–1157 (in Chinese with English abstract).
[14] 程立国. 大豆遗传图谱构建和重要性状的QTL定位.南京农业大学硕士学位论文, 江苏南京, 2008. Cheng L G. Construction of Genetic Linkage Map and QTL Mapping of Important Traits in Soybean [(L.) Merrill]. MS Thesis of Nanjing Agricultural University, Nanjing, Jiangsu, China, 2008 (in Chinese with English abstract).
[15] 丁卉. 利用SSR标记研究大豆对胞囊线虫抗性和主要农艺性状的自然选择效应. 南京农业大学硕士学位论文, 江苏南京, 2009. Ding H. The Natural Selection Effect on Resistance to SCN and Major Agronomic Characters of Soybean by SSR Analysis. MS Thesis of Nanjing Agricultural University, Nanjing, Jiangsu, China, 2009 (in Chinese with English abstract).
[16] 洪雪娟, 黄婧, 丁卉, 侯金锋, 李永春, 盖钧镒, 邢邯. 大豆异地衍生重组自交系群体产量相关性状的QTL定位. 中国油料作物学报, 2014, 36: 572–579. Hong H J, Huang J, Ding H, Hou J F, Li Y C, Gai J Y, Xing H. Detection of soybean QTLs on yield-related traits in RIL populations derived from Peking × 7605 in two sites.,2014, 36: 572–579 (in Chinese with English abstract).
[17] 袁道华. 大豆抗胞囊线虫基因遗传机制及育种价值分析. 河南农业大学硕士学位论文, 河南郑州, 2010. Yuan D H. Genetic Mechanisms and Breeding Value Analysis of Soybean Cyst Nematode Resistance Gene. MS Thesis of Henan Agricultural University, Zhengzhou, Henan, China, 2010 (in Chinese with English abstract).
[18] Sayama T, Hwang T Y, Yamazaki H, Yamaguchi N, Komatsu K, Takahashi M, Suzuki C, Miyoshi T, Tanaka Y, Xia Z J, Tsubokura Y, Watanabe S, Harada K, Funatsuki H, Ishimoto M. Mapping and comparison of quantitative trait loci for soybean branching phenotype in two locations., 2010, 60: 380–389.
[19] Yao D, Liu Z Z, Zhang J, Liu S Y, Qu J, Guan S Y, Pan L D, Wang D, Liu J W, Wang P W. Analysis of quantitative trait loci for main plant traits in soybean., 2015, 14: 6101–6109.
[20] 张霞. 大豆分枝相关基因的发掘与利用. 中国农业科学院硕士学位论文, 北京, 2017. Zhang X. Identification and Utilization of Genes Related to Branching in Soybean. MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2017 (in Chinese with English abstract).
[21] Shim S, Kim M Y, Ha J, Lee Y H, Lee S H. Identification of QTLs for branching in soybean [(L.) Merrill]., 2017, 213: 225–233.
[22] 谭冰, 郭勇, 邱丽娟. 大豆全基因组分枝相关基因发掘及与QTL共定位. 遗传, 2013, 35: 793–804. Tan B, Guo Y, Qiu L J. Whole genome discovery of genes related to branching and co-localization with QTLs in soybean.(Beijing), 2013, 35: 793–804 (in Chinese with English abstract).
[23] 邱丽娟. 大豆种质资源描述规范和数据标准. 北京: 中国农业出版社, 2006. p 22. Qiu L J. Description and Data Standards for Soybean [(L.) Merrill]. Beijing: China Agriculture Press, 2006. p 22 (in Chinese).
[24] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations., 2015, 3: 269–283.
[25] Yano M. Genetic and molecular dissection of naturally occurring variation., 2001, 4: 130–135.
[26] Ashikari M, Matsuoka M. Identification, isolation and pyramiding of quantitative trait loci for rice breeding., 2006, 11: 344–350.
[27] 鲁宁宁, 赵云雷, 王红梅, 陈伟, 赵佩, 龚海燕, 崔艳利, 桑晓慧, 张凯. 基于RIL群体鉴定棉花抗黄萎病相关QTLs. 棉花学报, 2019, 31: 254–262. Lu N N, Zhao Y L, Wang H M, Chen W, Zhao P, Gong H Y, Cui Y L, Sang X H, Zhang K. Identification of QTLs related to Verticillium wilt resistance based on RIL population., 2019, 31: 254–262 (in Chinese with English abstract).
[28] Kim K S , Diers B W, Hyten D L, Rouf Mian M A, Shannon J G, Nelson R L. Identification of positive yield QTL alleles from exotic soybean germplasm in two backcross populations., 2012, 125: 1353–1369.
[29] Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano L M, Kamoun S, Terauchi R. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations., 2013, 74: 174–183.
[30] 巩鹏涛, 李迪. 植物分枝发育的遗传控制. 分子植物育种, 2005, 3: 151–162. Gong P T, Li D. Genetic control of plant shoot branching., 2005, 3: 151–162 (in Chinese with English abstract).
[31] Kebrom T H, Spielmeyer W, Finnegan E J. Grasses provide new insights into regulation of shoot branching., 2013, 18: 41–48.
[32] Thimann K V, Skoog F. Studies on the growth hormone of plants: III. The inhibiting action of the growth substance on bud development., 1933, 19: 714–716.
[33] Patel S, Rose A, Meulia T, Dixit R, Cyr R J, Meier I.WPP-domain proteins are developmentally associated with the nuclear envelope and promote cell division., 2005, 16: 3260–3273.
[34] Cao L R, Lu X M, Zhang P Y, Wang G R, Wei L, Wang T C. Systematic analysis of differentially expressed maizegenes between drought and rewatering transcriptome reveals bZIP family members involved in abiotic stress responses., 2019, 20: 4103–4127.
[35] Riechmann J L, Heard J, Martin G, Reuber L, Jiang C Z, Keddie J, Adam L, Pineda O, Ratcliffe D J, Yu G.transcription factors: Genome-wide comparative analysis among eukaryotes., 2000, 290: 2105–2110.
[36] Jakoby M, Weisshaar B, Dröge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj T, Parcy F. bZIP transcription factors in., 2002, 7: 106–111.
[37] Nijhawan A, Jain M, Tyagi A K, Khurana J P. Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice., 2008, 146: 333–350.
[38] Silveira A B, Gauer L, Tomaz J P, Cardoso P R, Carmello- Guerreiro S, Vincentz M. Theprotein fused to the VP16 transcriptional activation domain alters leaf and vascular development.(Oxford), 2007, 172: 1148–1156.
[39] Fukazawa J, Sakai T, Ishida S, Yamaguchi I, Kamiya Y, Takahashi Y. Repressionr of shoot growth, a bZIP transcriptional activator, regulates cell elongation by controlling the level of gibberellins., 2000, 12: 901–915.
[40] Guan Y C, Ren H B, Xie H, Ma Z Y, Chen F. Identification and characterization of bZIP-type transcription factors involved in carrot (L.) somatic embryogenesis., 2009, 60: 207–217.
[41] Ulm R, Baumann A, Oravecz A, Mate Z, Adam E, Oakeley E J, Schafer E, Nagy F. Genome-wide analysis of gene expression reveals function of the bZIP transcription factor HY5 in the UV-B response of., 2004, 101: 1397–1402.
[42] Weltmeier F, Ehlert A, Mayer C S, Dietrich K, Wang X, Schutze K, Alonso R, Harter K, Vicente-Carbajosa J, Droge-Laser W. Combinatorial control ofproline dehydrogenase transcription by specific heterodimerisation of bZIP transcription factors., 2006, 25: 3133–3143.
[43] Kruger N J, Schaewen A V. The oxidative pentose phosphate pathway: Structure and organisation., 2003, 6: 236–246.
[44] Spielbauer G, Li L, Römisch M L, Do P T, Fouquet R, Fernie A R, Eisenreich W, Gierl A, Settles A M. Chloroplast- localized 6-phosphogluconate dehydrogenase is critical for maize endosperm starch accumulation., 2013, 64: 2231–2242.
Development of molecular markers and fine mapping oflocus related to branch number in soybean (L.)
WU Hai-Tao1,3,**, ZHANG Yong2,**, SU Bo-Hong1,3,**, Lamlom F Sobhi3,4, and QIU Li-Juan3,*
1College of Agriculture, Northeast Agricultural University, Harbin 150030, Heilongjiang, China;2Keshan Branch of Heilongjiang Academy of Agricultural Sciences, Qiqihar 161606, Heilongjiang, China;3National Key Facility for Gene Resources and Genetic Improvement / Key Laboratory of Crop Germplasm Utilization, Ministry of Agriculture / Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;4Plant Production Department, Faculty of Agriculture Saba Basha, Alexandria University, Alexandria, Egypt
The branch number is one of the important factors influencing soybean yield, which is directly related to pod setting rate. At the same time, it is also an important component of soybean plant type, and further affects the yield by adjusting the population structure and planting density. At present, there is few report related to map-based cloning of genes related to branch number. Therefore, the discovery of genes/QTL involved in the regulation of soybean branching is of great significance for the basic research on the establishment of plant type and the applied research on the development of high-yielding varieties. In this study, based on the F2of crossing low-branched variety Kenfeng 19 (KF19) and high-branched variety Kennong 24 (KN24), we developed the F7:8recombinant inbred line (RIL) population, consisting of 606 lines, and two backcrossing populations consisting of 1486 individuals for KF19-BC3F2and 1150 individuals for KN24-BC2F2. Within the localization interval of the new QTL of the branch number of chromosome 18 (), 11 polymorphism SSR markers were screened out to identify the RIL population, and region ofwas reduced from 1.6 Mb to 113 kb. After developing two InDel markers BR69 and BR77 in the mapping region, the backcross population was used to screen the exchange individuals, the interval ofwas further reduced to 63.7 kb, including 9 genes. Those results provide the information for gene map-based cloning and molecular marker assisted breeding of branch number in soybean.
soybean; branch number; QTL mapping; candidate genes
10.3724/SP.J.1006.2020.04043
本研究由“十三五”国家重点研发计划项目(2016YFD0100201), 中国农业科学院科技创新工程和大豆种质资源保护与利用项目(2019NWB036-05)资助。
The study was supported by the National Key Research and Development Program of China (2016YFD0100201), the Agricultural Science and Technology Innovation Program ofChinese Academy of Agricultural Sciences, and the Protection and Utilization of Soybean Germplasm Resources (2019NWB036-05).
邱丽娟, E-mail: qiulijuan@caas.cn
**同等贡献(Contributed equally to this work)
吴海涛, E-mail: 1960478192@qq.com
2020-02-25;
2020-06-02;
2020-06-22.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200622.1346.010.html
