高效液相色谱-串联质谱法测定盐酸二甲双胍及其制剂中痕量N-亚硝基二甲胺
2020-09-23郭常川杨书娟褚志杰徐玉文
郭常川, 刘 琦, 张 雷, 郑 静, 汪 勇, 杨书娟, 褚志杰, 牛 冲, 徐玉文*
(1. 山东省食品药品检验研究院, 山东 济南 250101; 2. 岛津企业管理(中国)有限公司, 北京 100020; 3. 山东省中医院, 山东 济南 250014)
N-亚硝基二甲胺(NDMA)又名N-二甲基亚硝胺,具有很强的肝毒性,ICH M7 (R1)指南明确指出该化合物具有较高的致癌性[1]。根据世界卫生组织公布的致癌物清单,NDMA属于2A类致癌物质[2]。自2018年以来,国内外多家药企都曾因缬沙坦、雷尼替丁产品中检出超限量的NDMA而陷入风波。2019年12月,美国食品药品监督管理局(FDA)开始调查已批准在美国销售的二甲双胍中的NDMA含量,并将酌情建议召回。随后,欧洲、加拿大、新加坡及我国药品监管部门也陆续对辖区内二甲双胍中NDMA的含量情况展开调查。2020年5月8日,国家药品监督管理局(NMPA)发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》。
目前,NDMA的检测方法主要有表面增强拉曼散射法(SERS)[3]、分子印迹聚合物电化学法[4]、HPLC-化学发光法[5]、液相色谱-质谱法(LC-MS)[6,7]、气相色谱-热能分析法[8]、气相色谱-质谱法(GC-MS)[8-13]等,主要应用于食品(饮用水、水产品、肉制品等)、化妆品和烟草行业。国内外关于药品中NDMA检测的研究较少,目前仅检索到缬沙坦、厄贝沙坦、吉非替尼中NDMA测定的文献[14-17],未见有针对二甲双胍中NDMA检测的报道。但测定其他药物中NDMA的文献不能直接应用于盐酸二甲双胍中NDMA的测定,这是因为药物主成分理化性质不同,导致提取方法、色谱保留均有差异,因此前处理方法、色谱条件不能通用,亟须重新开发方法。LC-MS由于其灵敏度高、选择性好的优势,在药物杂质或残留分析领域应用广泛[18]。本研究建立了用于测定盐酸二甲双胍原料药(API)及其制剂中NDMA含量的LC-MS法,样品前处理简便快速,灵敏度高,专属性强,重复性好,已用于盐酸二甲双胍API及其制剂中NDMA的检测。
1 实验部分
1.1 仪器、试剂与材料
超高效液相色谱-三重四极杆质谱系统包括LC-30AD高压二元液相泵、CBM-20A通讯总控模块、DGU-20A5R脱气机、SIL-30AC自动进样器、CTO-30A柱温箱以及LCMS-8050质谱仪,LabSolutions软件用于控制和数据处理(Shimadzu公司,日本); Mettler XS205型电子天平(Mettler Toledo公司,瑞士); Heraeus Multifuge X1R型高速冷冻离心机(Thermo Fisher Scientific公司,美国); KQ-500 DE型温控超声仪(昆山市超声仪器有限公司); THZ-82型水浴恒温振荡器(常州金坛精达仪器制造有限公司); 2 mL一次性无菌注射器(山东新华安得医疗用品有限公司); 13 mm、0.22 μm微孔滤头(上海安谱实验科技股份有限公司)。NDMA对照品购自梯希爱(上海)化成工业发展有限公司(含量>99.0%); HPLC级甲醇(纯度99.9%)、甲酸(纯度>99%)及超纯水均购自Thermo Fisher Scientific公司(美国)。86批盐酸二甲双胍原料药、16批盐酸二甲双胍片剂、11批二甲双胍格列本脲胶囊均为药企委托检验提供。
1.2 实验条件
1.2.1对照品溶液的制备
取NDMA对照品适量,精密称定,加水溶解并定量稀释为1.0 mg/mL的NDMA对照品溶液;以水作为稀释溶剂,逐级稀释得到1.0、2.0、5.0、10.0、50.0、100.0 ng/mL的系列NDMA工作溶液。
1.2.2样品前处理
取本品(原料、片、肠溶片)或本品内容物(胶囊)(约相当于盐酸二甲双胍500 mg),精密称定,置于50 mL离心管中,精密加入水10 mL,涡旋混匀1 min,再以350 r/min的频率振荡10 min,于10 000 r/min、4 ℃条件下离心5 min,取上清液过0.22 μm微孔滤膜,取续滤液即得(原料则无须过滤膜,取离心上清液即得)盐酸二甲双胍原料、片、肠溶片、胶囊供试品溶液。
1.2.3色谱条件
色谱柱:ACE EXCEL 3 C18-AR柱(150 mm×4.6 mm, 3 μm);柱温:40 ℃;自动进样器温度:10 ℃;流动相A:含0.1%甲酸的水溶液,流动相B:含0.1%甲酸的甲醇溶液;流速:0.8 mL/min。梯度洗脱程序:0~6.0 min, 5%B; 6.0~7.0 min, 5%B~90%B; 7.0~10.0 min, 90%B; 10.0~10.5 min, 90%B~5%B; 10.5~14.0 min, 5%B。进样量:10 μL。六通阀切换设置:保留时间2.85~7.00 min流动相进入质谱,其余时间流动相进入废液。
1.2.4质谱条件
离子源:大气压化学电离(APCI)源,正离子模式;监测方式:MRM模式;接口温度:300 ℃;脱溶剂管温度:250 ℃;加热块温度:400 ℃;雾化器流量:3 L/min;加热器流量:10 L/min;干燥器流量:10 L/min。
NDMA定量离子对为m/z75.0→43.1,驻留时间为197.0 ms,预四级杆偏转电压偏差Q1 Pre为-12.0 V,碰撞能量(CE)为-17.0 eV,预四级杆偏转电压偏差Q3 Pre为-46.0 V; NDMA定性离子对为m/z75.0→58.2,驻留时间为197.0 ms, Q1 Pre为-16.0 V, CE为-16.0 eV, Q3 Pre为-24.0 V。
1.3 检出量的计算
分别取对照品溶液和供试品溶液各10 μL注入液相色谱-串联质谱仪,按外标法计算供试品中NDMA含量。NDMA检出量(detected amount of NDMA,用ppm(10-6)表示)计算公式见公式1,
(1)
式中:Ai和As分别为供试品溶液和对照品溶液中NDMA的峰面积;CAPI为供试品溶液中盐酸二甲双胍的质量浓度(mg/mL);Cs为NDMA对照品溶液的质量浓度(mg/mL)。
2 结果与讨论
2.1 实验条件考察
2.1.1质谱条件优化
NDMA相对分子质量较小,母离子m/z仅为75.0。采用正离子模式进行质谱检测时,低质荷比(尤其是m/z<100)的离子通常基线噪声较大,影响色谱峰信噪比,造成低浓度检测的困难。而根据NMPA暂定限度要求折算,本实验NDMA定量限要低于1.60 ng/mL,且S/N≥10,对仪器灵敏度要求很高。因此,为实现NDMA的痕量检测,本研究通过母离子扫描、子离子扫描等方式筛选出多组NDMA多反应监测离子对,再应用Labsolution软件优化选择出最佳离子对、碰撞能量及驻留时间,并对离子源气流速、温度、电压进行详尽优化,最终实现了NDMA检测的灵敏度要求。
2.1.2色谱条件优化
样品溶液中含有高含量的二甲双胍(50.00 mg/mL)和痕量的NDMA(<100.00 ng/mL),在进行HPLC-MS检测时,如果高含量二甲双胍进入质谱系统,会污染离子源并产生严重的基质效应,会对NDMA测定造成很大影响。因此,应使NDMA和二甲双胍达到足够的色谱分离度。实验通过六通阀将二甲双胍色谱峰处的流动相切换到废液,避免高含量二甲双胍进入质谱。
本实验最初考察100 mm×4.6 mm规格色谱柱,流速为0.5 mL/min,发现NDMA和二甲双胍色谱分离较差,保留时间仅相隔1.2 min,对阀切换造成了困难。改用150 mm×4.6 mm规格色谱柱,流速增大为0.8 mL/min,优化梯度洗脱条件使NDMA和二甲双胍实现了较好的色谱分离。二甲双胍色谱峰保留时间为2.24 min,出峰较早,于2.60 min左右回到基线,NDMA色谱峰保留时间约为4.40 min,通过六通阀控制仅将保留时间2.85~7.00 min的流动相进入质谱系统,有效避免了高含量API的基质干扰,保护了离子源,使NDMA的测定更加准确可靠。
此外,为进一步提高灵敏度,本实验考察了进样体积的影响,在不影响峰形的前提下,尽量增大进样体积。实验发现进样体积大于10 μL时NDMA峰形开始变差,因此本实验选定进样量为10 μL。
2.1.3前处理方法优化
NDMA易溶于水、醇,但二甲双胍在水中易溶,在甲醇中溶解。实验发现,如果选用甲醇作为提取溶剂,则供试品溶液在自动进样器10 ℃条件下放置1 h后即发生结晶析出的现象,系二甲双胍在甲醇中过饱和所致,会堵塞进样器和色谱柱。因此,本实验选用水作为提取溶剂,使NDMA和二甲双胍均能形成澄清透明的水溶液,再进行超声提取、微孔滤过操作,前处理简单,重复性好,回收率高。
2.2 方法学考察
2.2.1专属性
将水、处理后的辅料溶液、NDMA对照品溶液、处理后的样品(批号18120201)溶液分别进样测定(见图1)。结果表明,NDMA出峰时间约为4.40 min,相同保留时间处的溶剂及辅料溶液均无干扰峰,方法专属性良好。
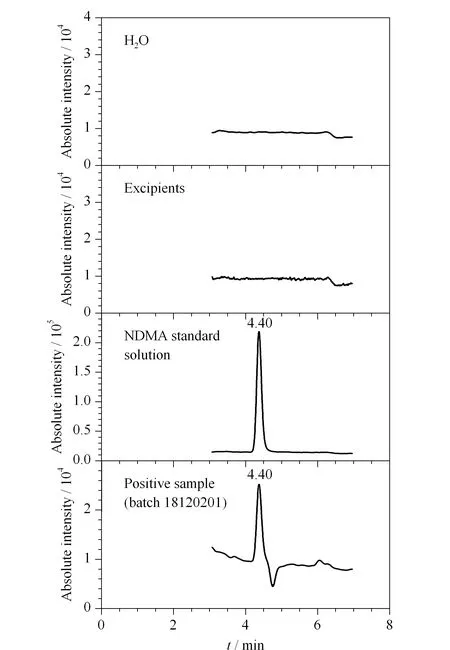
图 1 水、辅料、NDMA及阳性样品提取离子色谱图Fig. 1 Extracted ion chromatograms of H2O, excipients, NDMA and a positive sampleNDMA: N-dimethylnitrosamine.
2.2.2线性范围
取系列标准溶液进样测定。以峰面积作为y, NDMA标准溶液质量浓度(ng/mL)作为x,不加权重拟合线性校正曲线,建立回归方程,计算相关系数(r)。结果表明,NDMA在1.00~100.00 ng/mL范围内线性关系良好,线性回归方程为y=20 232.50x-871.25,r=0.999 97。
2.2.3检出限和定量限
精密移取1.00 ng/mL NDMA标准溶液,以水作为溶剂逐级稀释,按信噪比S/N≥ 3和10计算检出限和定量限。结果表明,方法的检出限为0.20 ng/mL,定量限为1.00 ng/mL。
2.2.4回收率和精密度
根据NMPA对亚硝胺类杂质限度要求以及盐酸二甲双胍说明书中规定的每日最大用药量计算,盐酸二甲双胍原料含NDMA不得过0.032 ppm,盐酸二甲双胍制剂含NDMA不得过0.048 ppm[19]。本研究考察了低、中、高3个水平的NDMA在盐酸二甲双胍原料药及制剂中的回收率和精密度。取已知阴性样品约一次服用量(以盐酸二甲双胍计500.00 mg),原料药及制剂皆精密称取9份(低、中、高水平各3份),先分别精密加入适量的NDMA对照品溶液,后续操作按照1.2.2节描述处理,得到加标回收试验用供试品溶液。结果表明,低、中、高3个水平浓度下的NDMA在盐酸二甲双胍原料药及制剂中的加标回收率为94.55%~114.67%, RSD为4.73%~13.46%(见表1),符合药典规定,表明NDMA回收率和重复性良好。

表 1 不同剂型中NDMA的加标回收率(n=3)
2.2.5基质效应
分别制备低、中、高3个水平的空白基质加标溶液和对照品标准溶液,分别进样,计算空白基质加标溶液和对照品标准溶液中NDMA峰面积之比,即为基质效应。NDMA在低、中、高3个水平下的基质效应为88.73%~103.61%,表明空白基质不影响NDMA的质谱测定。
2.2.6稳定性
将1.00 ng/mL的NDMA标准溶液保存于进样瓶中,在自动进样器中按照实验条件放置0、8、24 h后进样测定,NDMA峰面积的RSD为2.08%,说明该条件下NDMA在24 h内稳定性良好。
2.3 实际样品检测
对113批盐酸二甲双胍原料药及制剂处理后进样测定,外标法计算样品中NDMA检出量。根据NMPA制定的NDMA限度要求[19],原料药NDMA含量均不超限,但有8批二甲双胍制剂(含2批片剂、6批胶囊)超过了限度,其质量浓度为3.59~14.10 ng/mL,检出量为0.072~0.282 ppm。阳性样品测定结果见表2。

表 2 阳性样品测定结果
3 结论
本研究建立了盐酸二甲双胍中NDMA的HPLC-MS检测方法,并进行了详尽的方法学验证,同时将方法应用于盐酸二甲双胍原料药及制剂中NDMA的测定。本方法灵敏度高,专属性好,回收率高,线性范围宽,操作简便、可靠,已用于盐酸二甲双胍原料药及制剂中NDMA检验工作,填补了该领域研究空白。本研究可以帮助企业进行生产工艺控制,并为药监部门的监管提供有力的技术支持。
