热可逆交联等规聚丙烯的合成
2020-09-15刘小燕
包 卓,刘小燕,牛 慧,李 杨
(1. 大连理工大学 化工学院 高分子材料系,辽宁 大连 116024;2. 中国石油 石油化工研究院 兰州化工研究中心,甘肃 兰州 730060)
等规聚丙烯是重要的通用树脂,具有密度低、熔点高、力学性能优、化学稳定性好等特点,在汽车、家电、建筑、居民日常生活领域都有广泛应用[1]。特别是近年来,受单体来源多样化的推动,国际国内聚丙烯产能井喷式增长,应用领域不断拓展,也对聚丙烯性能提出了更高的要求。提高聚丙烯分子量可以显著增强材料的力学性能,但却使其加工性变差(如熔体流动性减弱,加工效率降低等)。交联是对材料进行增强的有效手段,包括有机过氧化物交联和辐照交联等方法[2-4]都被应用于聚丙烯的增强,但交联聚丙烯是一类热固性材料,一旦成型就难以进行再次熔融加工,这给加工制造和回收利用带来诸多限制[5]。长久以来,聚丙烯的力学性能和加工性能始终是一对矛盾。目前工业上主要通过加宽聚丙烯分子量分布以实现加工性能与力学性能的平衡:低分子量聚合物增加熔体流动性,高分子量聚合物增加分子链缠结进而提高材料的力学性能。
呋喃/马来酰亚胺之间的Diels-Alder(D-A)反应([4+2]环化反应)具有原料易得、易发生逆向D-A反应(rD-A)的特点。在常温或加热下(60℃左右)呋喃/马来酰亚胺之间发生D-A反应,在120 ℃左右发生rD-A反应,是获得热可逆结构的一种有效途径[6-11]。如能利用该反应实现聚丙烯分子链之间的化学交联,同时利用该化学键在120 ℃以上断开从而使聚丙烯解交联的特性,就能获得低温强化(交联)、高温高流动(解交联)的新型聚丙烯树脂,为聚丙烯力学性能和加工性能的平衡提供全新的解决方案。
本工作通过设计新型功能性单体6-呋喃-1-己烯(fH),在等规聚丙烯侧基上引入呋喃取代基,再以双马来酰亚胺小分子为交联剂,利用呋喃/马来酰亚胺之间的D-A及rD-A反应,制备了可逆交联等规聚丙烯。采用1H NMR,13C NMR,DSC,FTIR,XRD等方法分析了fH对聚合动力学、聚合竞聚率及聚合物性能的影响,并考察了可逆交联等规聚丙烯的力学性能。
1 实验部分
1.1 主要原料
呋喃:分析纯,上海麦克林生物化学技术有限公司,含0.025%(w)2,6-二叔丁基对甲酚稳定剂,分子筛除水后使用;6-溴-1-己烯:纯度98%,萨恩化学技术(上海)有限公司,分子筛除水后使用;正丁基锂、三乙基铝:分析纯,阿拉丁试剂公司;四氢呋喃(THF):分析纯,天津市富宇精细化工有限公司,经钠蒸馏除水后使用;丙烯:聚合级,大连光明特种气体有限公司;1-己烯:纯度98%,经氢化钙搅拌24 h除水后减压蒸馏使用,安耐吉试剂公司;正己烷:分析纯,天津市大茂试剂厂,氮气气氛下用金属钠回流除水后蒸出使用。
CS-1型催化剂:Ziegler-Nata催化剂,营口向阳催化剂厂。
1.2 fH的制备
在氮气保护下,向250 mL单口瓶中加入无水无氧50 mL THF作溶剂,加入10.02 g呋喃,用液氮和乙醇做冷浴,保持-80~-70 ℃下逐滴滴入1.6 mol/L的正丁基锂89.6 mmol,室温反应4 h;然后在氮气保护中,保持-80~-70 ℃下逐滴滴入混合均匀的6-溴-1-己烯126 mmol与20 mL THF,室温反应过夜;撤掉氮气,用去离子水和乙酸乙酯进行萃取,所得油相经无水硫酸钠除水后旋蒸,得到的产物再用氢化钙除水,最后通过减压蒸馏得到fH约7 mL。
1.3 聚合反应
常温下向干燥的250 mL 圆底烧瓶中通入丙烯气体,用注射器依次加入50 mL正己烷、fH、外给电子体二环戊基(二甲氧基)硅烷、三乙基铝,搅拌5 min后再用20 mL正己烷将Ziegler-Natta催化剂冲50 mg入反应瓶中,45 ℃恒温进行聚合反应,聚合反应过程中向反应瓶内持续通入丙烯气体以保证聚合压力恒定为0.1 MPa。反应结束后将聚合产物倒入酸醇(盐酸/乙醇体积比为1∶4)溶液中终止反应,然后依次用乙醇、蒸馏水洗涤聚合产物,最后在40 ℃下真空干燥12 h后得到聚合产物。
1.4 交联反应
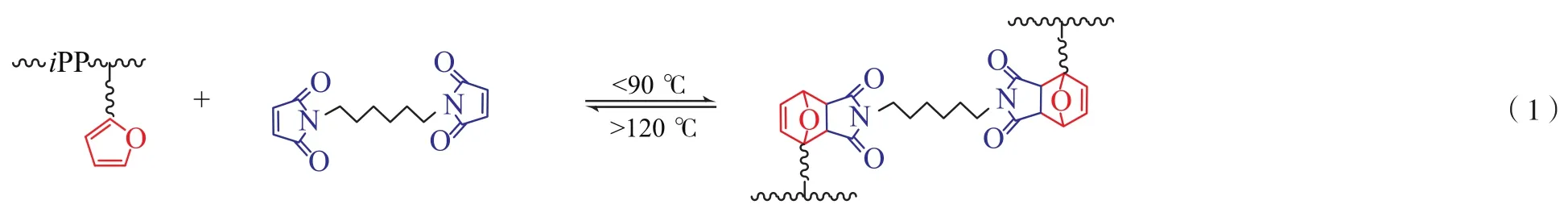
以二甲苯作溶剂,115 ℃下将聚丙烯溶于二甲苯溶液中,搅拌30 min后,降温至75 ℃,加入双马来酰亚胺交联剂(合成方法参照文献[12]),搅拌反应24 h。反应结束后将混合物倒入甲醇中沉降,依次用甲醇和去离子水洗涤产物后在40 ℃下真空干燥12 h后得到交联产物,交联反应原理见式(1)。
1.5 表征与测试
1H NMR和13C NMR表征采用Varian公司Bruker-400型核磁共振波谱仪,以氘代邻二氯苯为溶剂在100 ℃下进行检测。聚合物的结晶熔融过程采用TA公司Q2000型示差扫描量热仪测试,N2氛围下,以10 ℃/min的速率从50 ℃升温至200℃,恒温5 min以消除热历史;再以10 ℃/min的速率降温至-60 ℃并恒温2 min后,以10 ℃/min的速率升温至200 ℃。FTIR测试采用透射法在布鲁克公司6700型高级傅里叶变换红外光谱仪上进行,扫描次数32。XRD测试采用日本理学公司D/MAX 2400型全自动X射线粉末衍射仪。聚合物的拉伸性能采用Instron公司5567A型拉伸试验仪测定,起始长度5 mm,室温,湿度25%,拉伸速率50 mm/min。
2 结果与讨论
2.1 丙烯/fH共聚反应
采用CS-1型催化剂催化丙烯与fH共聚,通过调整fH的单体浓度,得到了呋喃取代基含量递变的等规聚丙烯,聚合条件和实验结果见表1。从表1可看出,单体fH用量对共聚反应活性影响不大。
聚合产物的1H NMR谱图见图1。从图1可看出,化学位移δ在0.95~1.65处的3个主峰是聚丙烯的特征峰;δ在2.60处的吸收峰归属于与呋喃环相连的亚甲基(a);δ在5.93,6.19,7.22处的3个特征峰归属于呋喃环上的氢(b~d)。从图中可明显观察到fH成功共聚进入聚丙烯链中,聚合物中的呋喃含量随聚合体系中fH用量的增加而升高(特征峰强度逐渐增加)。结合表1可知,当fH用量从0.05 mol/L增至0.20 mol/L时,fH含量从1.28%(x)增加到3.96%(x),转化率则有所下降,从56%降至42%。
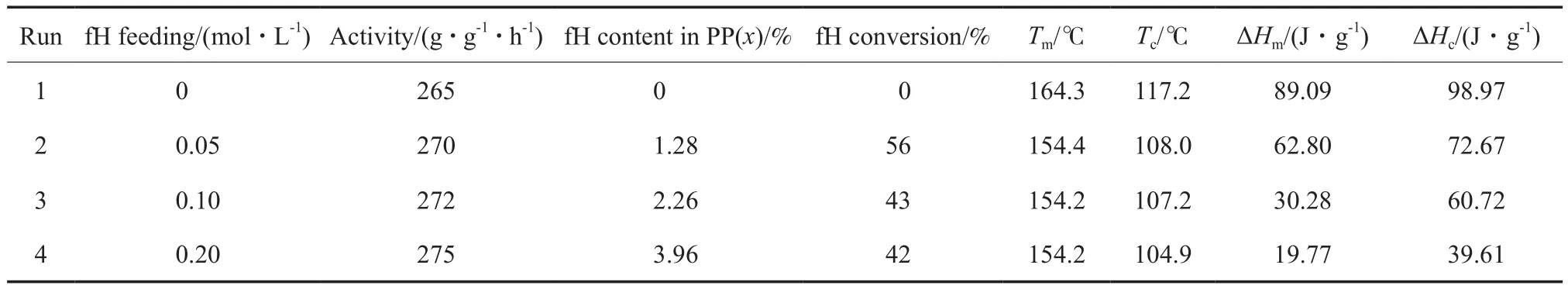
表 1 丙烯/fH共聚合反应及聚合物性能Table 1 Copolymerization of propylene/6-furyl-1-hexene(fH)
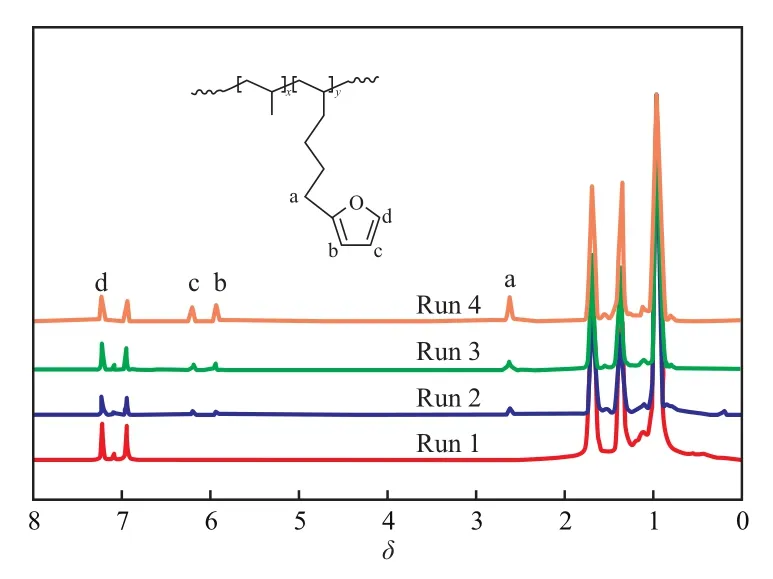
图1 丙烯/fH共聚物的1H NMR 谱图Fig.1 1H NMR spectra of propylene/fH copolymers.
考察了丙烯/fH共聚反应的动力学,并与不加入fH的丙烯聚合体系对比,结果见图2。从图2可看出,随聚合时间的延长,丙烯聚合活性呈先增加后降低,最后缓慢衰减的趋势,且在60 min左右聚合结束时仍然保持着一定的活性。添加了fH单体的聚合体系在聚合初期的活性高于不加入fH的丙烯均聚反应,这可能是由于fH在聚丙烯中的插入在一定程度上抑制了聚合过程中等规聚丙烯的结晶,有助于单体的扩散所致[13]。丙烯/fH共聚曲线位于丙烯均聚曲线下方,说明加入fH后聚合体系的活性整体上略低于丙烯均聚的活性,这与表1中的聚合活性数据不符。考虑到表1中实施的聚合时间为30 min,而图2显示出丙烯/fH共聚活性主要是在聚合进行到30 min左右才呈明显下降趋势。因此,fH对聚合活性中心的影响还需要进一步深入研究。
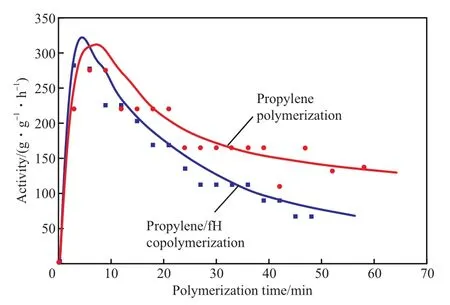
图2 丙烯(共)聚合反应动力学曲线Fig.2 Kinetic profiles of propylene (co)polymerization.
单体在增长链活性中心上的增长方式包括如下四种可能:

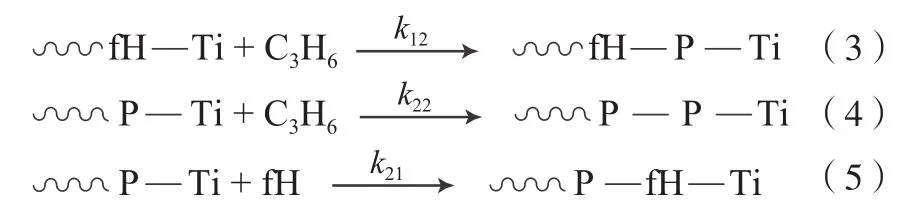
式中,P为丙烯;k为链增长速率常数;下标1为共聚单体,2为丙烯。
测定单体在某一浓度下的共聚物组成(为保证单体浓度在反应过程中近似恒定,需要选取低转化率下的数据进行计算),计算f和F(f和F分别为反应物和聚合物中的共聚单体与单体总量的摩尔比),根据Fineman-Ross方程(F-1)/f=r1–r2(F/f2)作图,通过截距可求出r1(r1=k11/k12),通过斜率可求出r2(r2=k22/k21)。
为研究fH中的呋喃取代基对共聚反应的影响,考察了fH与丙烯共聚的竞聚率,并与不含呋喃取代基的1-己烯进行对比。图3分别给出了丙烯/1-己烯和丙烯/fH共聚的Fineman-Ross曲线。从图3可看出,在丙烯/1-己烯共聚反应中,己烯的竞聚率为0.028;而在丙烯/fH共聚反应中,fH的竞聚率为0.045;丙烯的竞聚率变化不大。可见,尽管呋喃取代基具有一定的极性,但对于另一端的α-烯烃(1-己烯基)聚合影响不大,这可能是由于呋喃基与双键之间相隔较远,不易对活性中心产生影响;此外,fH的竞聚率相对1-己烯有所提高,这可能是由于位阻更大的fH在聚丙烯中的插入对聚合过程中等规聚丙烯结晶的抑制程度比1-己烯更明显,更有助于fH单体的扩散,这与前述的聚合动力学曲线特征分析结果一致。
聚合产物的DSC曲线见图4。从图4可看出,共聚单体fH在聚丙烯链中的插入对聚合物的熔点和结晶温度均有明显影响。与丙烯均聚物(Run 1)相比,少量fH的插入就可使共聚物的熔点从164℃降至154 ℃,熔融焓随fH含量的增加而不断减小,但熔点并不随着fH含量的增加而持续下降,Run2~4产物的熔点均为154 ℃左右;结晶温度和结晶焓的变化也有相似规律。fH的位阻效应是共聚物熔融焓降低(结晶度减小)的主要原因。
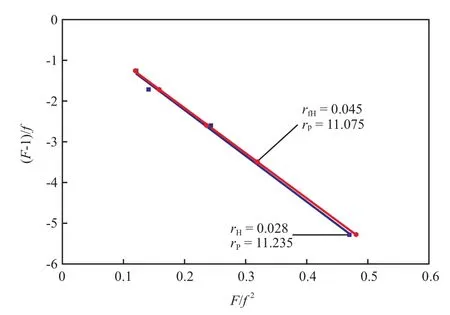
图3 基于Fineman-Ross方程计算单体竞聚率Fig.3 Calculation of reactivity ratio determined by Fineman-Rose equation.

图4 共聚物的DSC 熔融(a)和结晶(b)曲线Fig.4 DSC melting(a) and crystallization(b) curves of copolymers.
2.2 可逆交联聚丙烯的制备
采用fH含量分别为2.26%(x)和3.96%(x)的聚合产物(分别记为A0,B0),与双马来酰亚胺小分子(交联剂)进行交联反应,控制马来酰亚胺基与呋喃基的摩尔比为0.5,所得交联产物分别记为A1,B1,交联产物在180 ℃下可熔融热压成型。
交联前后试样的FTIR谱图见图5。从图5可看出,1 197 cm-1处的特征峰归属于D-A反应形成的环状分子的面内弯曲振动[14],交联后的试样A1中1 197 cm-1处的吸收峰强度增大,证实了D-A反应在聚丙烯中实施的可行性。
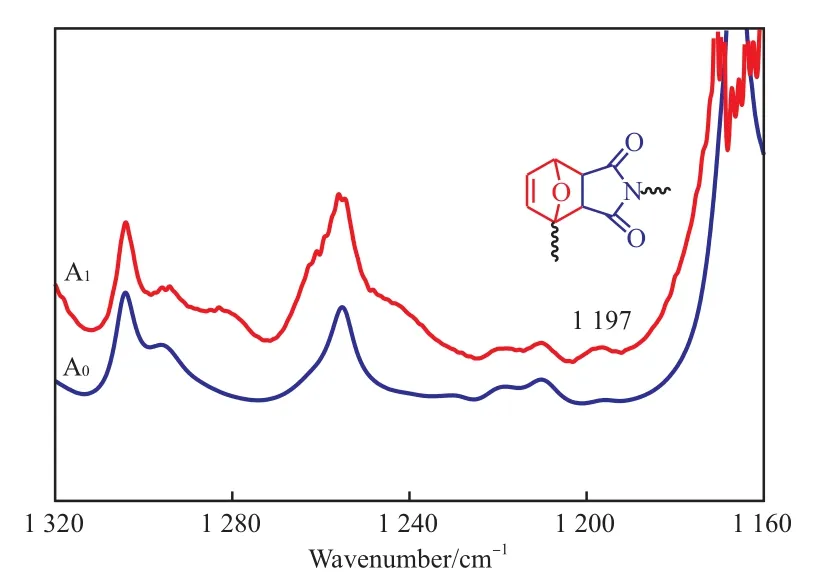
图5 交联前后试样的FTIR谱图Fig.5 FTIR spectra of the samples before and after cross-linking.
交联前后的试样均可进行热压成型。试样拉伸的应力应变曲线见图6。从图6可看出,未交联的A0,B0的拉伸强度、断裂伸长率、杨氏模量等均低于交联后的A1,B1,且fH含量更高的B0性能最差,这一方面是由于聚合物结晶度的下降导致材料刚性削弱,另一方面由于聚丙烯大分子之间没有足够缠结,拉伸直接导致材料破坏(甚至未出现屈服)。A1和B1的拉伸曲线均表现出显著的屈服和应变强化现象,断裂伸长率和断裂应力明显增加,说明在聚丙烯中引入交联能显著提升材料性能。
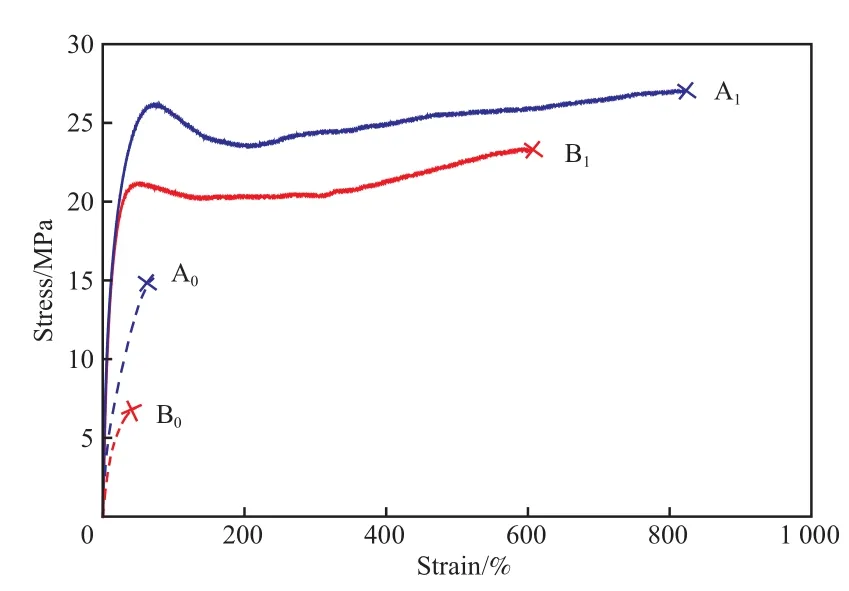
图6 聚合物的应力-应变曲线Fig.6 Stress-strain curves of polymers.
进一步利用XRD考察了fH单体的引入和可逆交联键对聚合物晶型的影响。试样交联前后的XRD谱图见图7。从图7可看出,2θ=14.1°,16.8°,18.6°处的吸收峰分别对应于等规聚丙烯(110),(040),(130)晶面,为聚丙烯的α-晶型特征峰[14],交联前后试样的峰形、位置及大小并无明显变化,说明采用fH和小分子交联剂(双马来酰亚胺)对聚丙烯的晶型无影响。
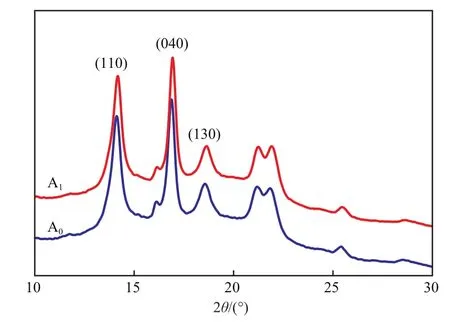
图7 聚合物的XRD 谱图Fig.7 XRD spectra of the polymers.
3 结论
1)通过Ziegler-Natta催化剂催化丙烯与功能性单体fH共聚,得到含呋喃侧基的等规聚丙烯,fH中的极性呋喃取代基对共聚活性影响不大,随fH用量的增大,fH含量增大,但转化率下降。
2)fH的位阻效应使丙烯/fH共聚物的熔点和结晶度较丙烯均聚物低。
3)以双马来酰亚胺为交联剂与丙烯/fH共聚物进行交联,可得到热可逆交联聚丙烯,交联后聚丙烯的杨氏模量、断裂伸长率及断裂应力均有显著提高,且fH和双马来酰亚胺的加入对聚丙烯的晶型无影响。
